





























We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
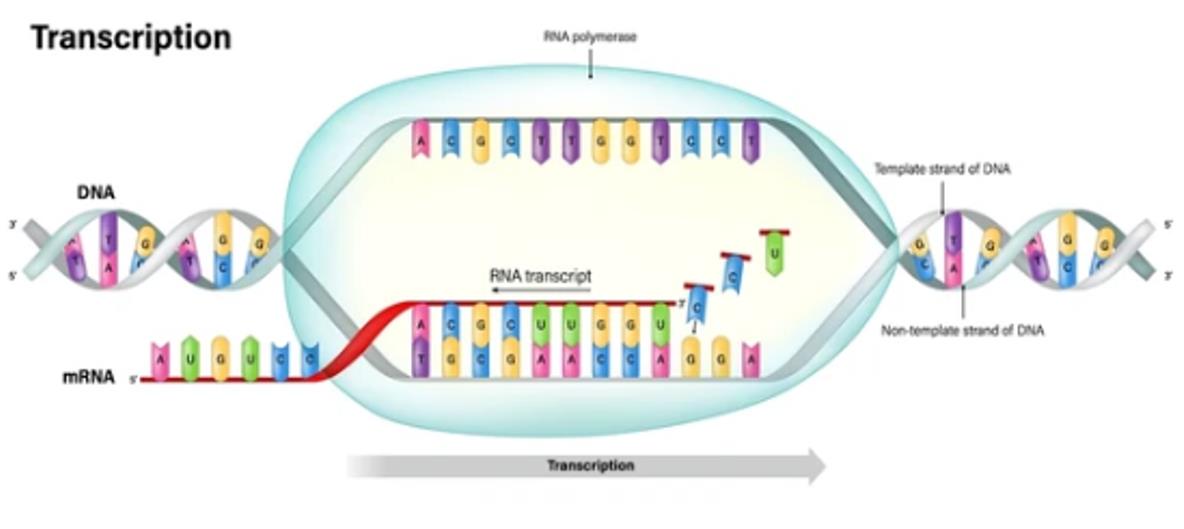
RESOURCE
For Research Use Only. Not for use in diagnostic procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment