We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Applications of Iso-Seq in the Field of Agriculture
At a glance:
- Iso-Seq Helps Elucidate the Regulatory Mechanisms of Growth and Development
- Iso-Seq Helps Analyze Stress and Resistance Regulation Mechanisms
- Iso-Seq Helps Identify Biosynthetic and Metabolic Mechanisms
- Iso-Seq Helps Build and Improve Reference Gene Sets
- Conclusion
Isoform Sequencing (Iso-Seq) is a powerful third-generation sequencing technology that provides full-length transcript sequences, offering unprecedented insights into the complexity of plant transcriptomes. This technology has revolutionized agricultural research by enabling detailed studies of gene expression, alternative splicing, and novel gene discovery. Here are some key applications of Iso-Seq in the field of agriculture.
Iso-Seq Helps Elucidate the Regulatory Mechanisms of Growth and Development
Studying the dynamic changes of the transcriptome at different developmental stages can accurately identify changes in transcript structure and expression of genes or transcripts at different stages, which helps to clarify the regulatory mechanism of plant growth and development.
An article published in Int J Mol Sci (IF 4.9) in 2022 performed isoform sequencing (Iso-Seq) on soybean underground tissues inoculated and not inoculated with rhizobia, and obtained 200,681 full-length transcripts covering 26,183 gene loci. The results showed that 80.78% of multi-exon sites produced more than one splicing variation. Comprehensive analysis found that during nodule development, especially in defense and transport-related processes, 7,874 different splicing events had highly diverse splicing patterns. The study further analyzed the genes used by different isoforms and revealed that 2,008 multi-subtype sites underwent stage-specific or simultaneous major subtype conversion after rhizobia inoculation, indicating that alternative splicing (AS) is an important way to regulate rhizobia development. This study was the first to identify 1,563 high-confidence long non-coding RNAs (lncRNAs) in soybean, of which 157 were differentially expressed during nodule development. The study revealed the landscape of AS during the interaction between soybean and rhizobia, providing systematic transcriptomic data for future research in multiple new directions of soybean.
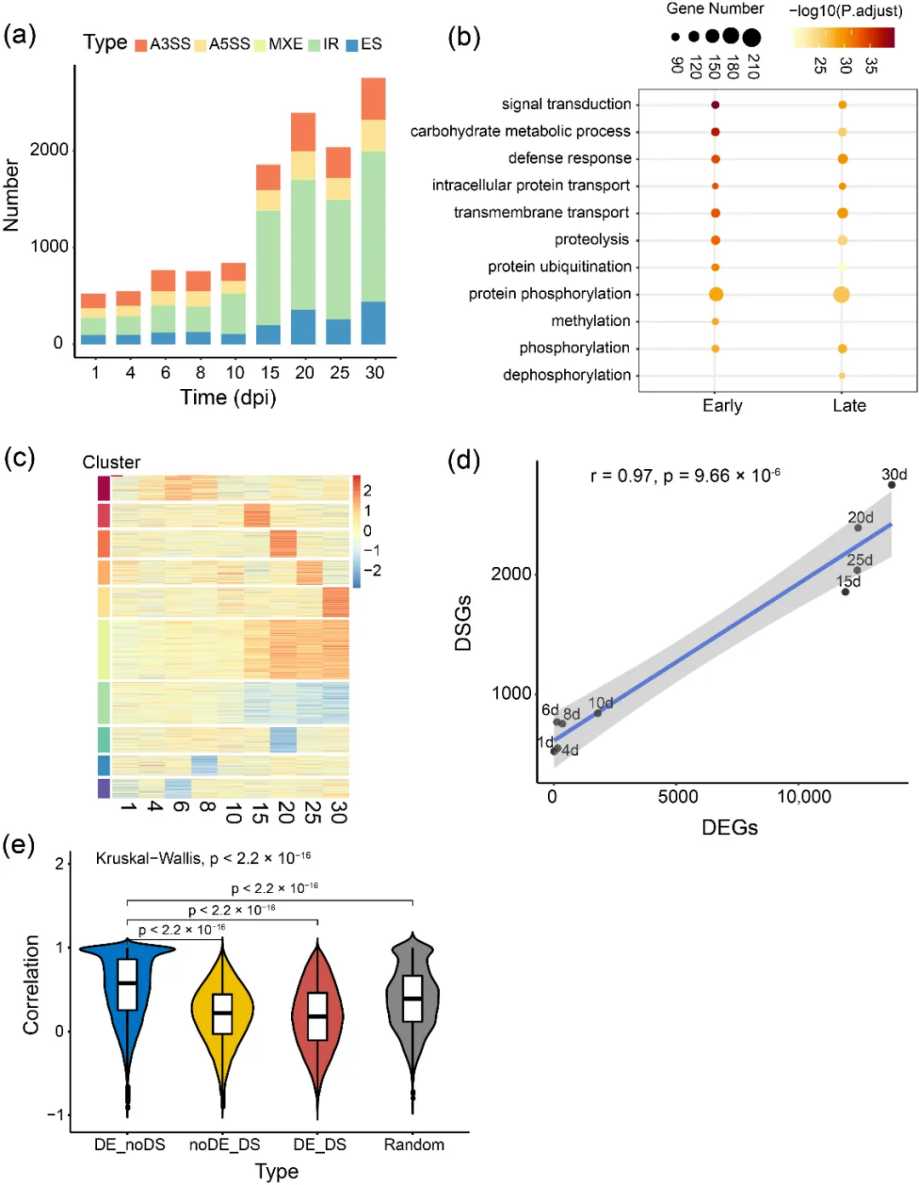 Figure 1. Genome-wide analysis of alternative splicing dynamics during nodule development.(Liu, J et al. 2022)
Figure 1. Genome-wide analysis of alternative splicing dynamics during nodule development.(Liu, J et al. 2022)
Iso-Seq Helps Analyze Stress and Resistance Regulation Mechanisms
Screening of differentially expressed genes/transcripts related to stress and resistance, combined with pathway enrichment analysis, can increase the understanding of complex regulatory networks related to stress adaptation and tolerance.
The extensive natural variation present in rice is an important source of genes to promote stress tolerance breeding. However, most stress-tolerant varieties lack high-quality genome assemblies. In an article published in Int J Mol Sci (IF 4.9) in 2020, the transcriptomes of ten rice varieties from three subspecies were sequenced on the PacBio platform. RNA was isolated from different organs of plants grown under control and abiotic stress conditions in different environments. The constructed reference transcriptome produced 37,500 to 54,600 plant-specific high-quality isoforms per variety. About 40% of the identified transcripts were new isoforms compared to the Nipponbare reference transcriptome. For the drought/heat tolerant variety N22, 56 differentially expressed genes were identified in developing seeds under combined field conditions of high temperature and drought. The newly generated rice transcriptome can be used to identify candidate genes for stress tolerance breeding that are not present in the reference transcriptome/genome. In addition, this method provides a cost-effective alternative to genome sequencing for identifying candidate genes in high stress tolerance genotypes.
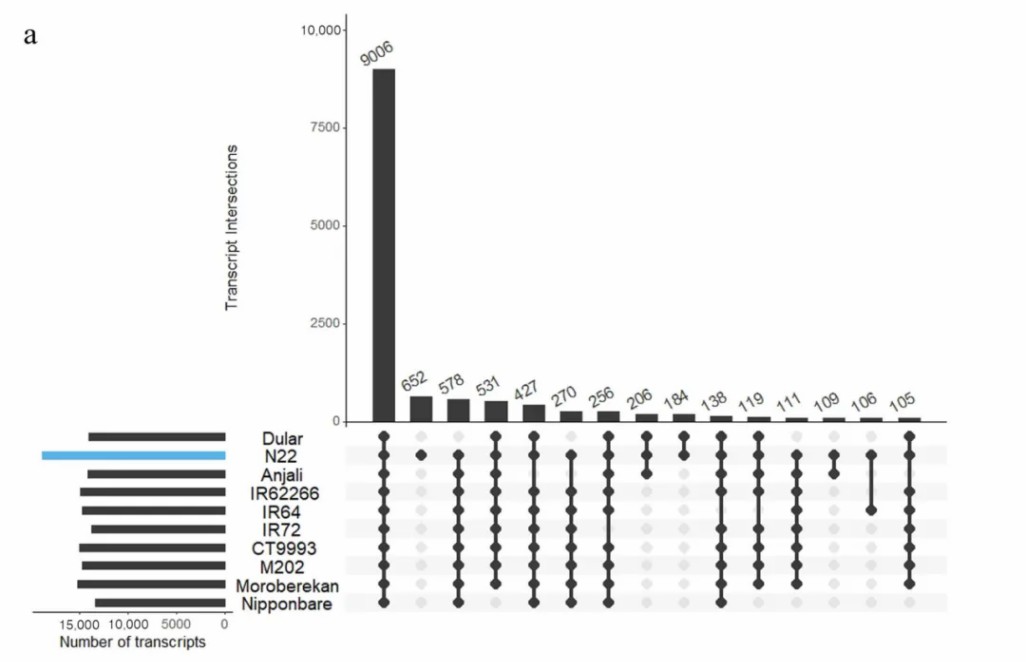 Figure 2. Identification of common and specific transcripts of all varieties.(Schaarschmidt et al. 2020)
Figure 2. Identification of common and specific transcripts of all varieties.(Schaarschmidt et al. 2020)
Iso-Seq Helps Identify Biosynthetic and Metabolic Mechanisms
Study biosynthesis or metabolic mechanisms, identify complete transcripts, and conduct in-depth research on key genes or transcripts of important metabolic pathways, breaking through the limitation of previous studies on the biosynthesis of important secondary metabolites of organisms using second-generation sequencing technology that could not accurately predict splicing isoforms.
Many species of the Urticaceae family are important cultivated fiber plants, and their economic and industrial value is well known. However, their secondary metabolite profiles and related biosynthetic mechanisms have not been well studied. An article published in J Agric Food Chem (IF 6.1) in 2024 used Laportea bulbifera as a model and conducted an extensive targeted metabolomics study, discovering 523 secondary metabolites, including unique flavonol glycoside accumulation in bulbils. In addition, the study identified relevant genes in the flavonoid biosynthetic pathway through full-length transcriptome and RNA-seq analysis. Finally, through weighted gene correlation network analysis and functional characterization, the study found that four LbUGTs (Glycosyltransferases of Artemisia ulmoides), including LbUGT78AE1, LbUGT72CT1, LbUGT71BX1 and LbUGT71BX2, can catalyze the glycosylation of flavonol aglycones (kaempferol, myricetin, gossypol and quercetin) with UDP-Gal and UDP-Glu as sugar donors. LbUGT78AE1 and LbUGT72CT1 showed substrate heterozygosity, while LbUGT71BX1 and LbUGT71BX2 showed different substrate and sugar donor selectivity. These results provide genetic resources for studying Artemisia ulmoides, a plant of the Urticaceae family, and key enzymes responsible for the metabolism of precious flavonoid glycosides.
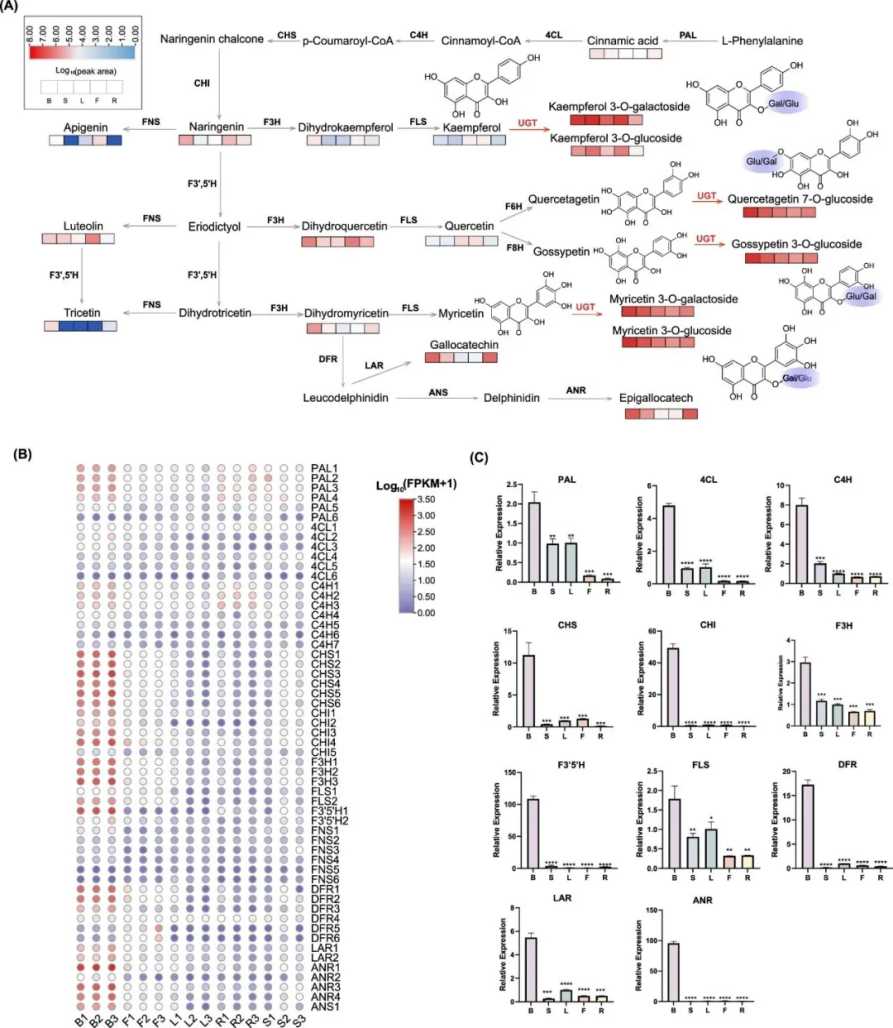 Figure 3. Identification and analysis of genes related to flavonoid biosynthesis.(Wang, W et al. 2024)
Figure 3. Identification and analysis of genes related to flavonoid biosynthesis.(Wang, W et al. 2024)
Iso-Seq Helps Build and Improve Reference Gene Sets
For species with references, we can improve the reference gene set and enhance the genome annotation. For species without references, we can build a high-quality gene set to lay the foundation for subsequent functional research of species.
As an important pollinator, bees play a vital role in maintaining ecological balance and providing products for humans. Although several versions of the Western honey bee genome have been published, its transcriptome information still needs to be improved. The article published in Int J Mol Sci (IF 4.9) in 2023 used PacBio single-molecule sequencing technology to sequence the full-length transcriptome of mixed samples from multiple developmental time points and tissues of queens, workers and drones of Melifora. A total of 116,535 transcripts were obtained, corresponding to 30,045 genes. Among them, 92,477 transcripts were annotated. Compared with the annotated genes and transcripts on the reference genome, 18,915 gene loci and 96,176 transcripts were newly identified. From these transcripts, 136,554 alternative splicing (AS) events, 23,376 alternative polyadenylation (APA) sites, and 21,813 lncRNAs were detected. In addition, many differentially expressed transcripts (DETs) between queen bees, worker bees, and drones were identified based on full-length transcripts. The results of this study provide a complete set of reference transcripts for the honey bee transcriptome, greatly expanding the understanding of the complexity and diversity of the honey bee transcriptome.
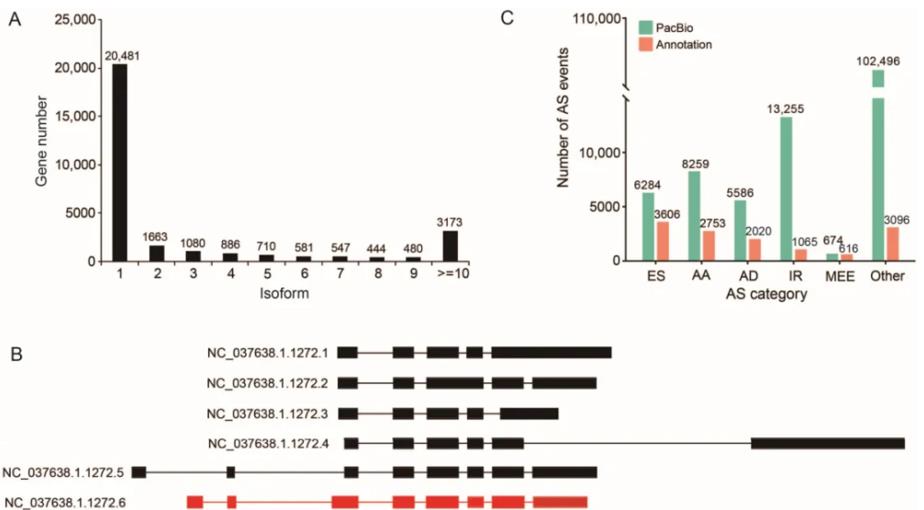 Figure 4. Variable splicing events identified from the PacBio bee transcriptome.(Zheng, S. Y et al. 2023)
Figure 4. Variable splicing events identified from the PacBio bee transcriptome.(Zheng, S. Y et al. 2023)
Conclusion
Iso-Seq is an advanced third-generation sequencing technology that provides full-length transcript sequences and has a wide range of applications in agricultural research. This technology can analyze the dynamic changes of the transcriptome in plant growth and development, such as the study of soybean rhizobium symbiosis, which revealed the key role of variable splicing in nodule development. In addition, Iso-Seq helps to analyze plant stress resistance mechanisms, such as the screening of tolerance genes for drought- and heat-resistant rice varieties, and provides candidate genes for stress-resistant breeding. This technology also supports the biosynthesis of secondary metabolites, such as the identification of key enzymes for flavonoids in pearl buds. In addition, Iso-Seq can be used to improve reference gene sets, such as the study of honey bee transcriptomes, which revealed a large number of new gene loci and transcripts, expanding the understanding of their gene regulation. Overall, the application of Iso-Seq in agriculture has promoted crop improvement, stress resistance research, and metabolic mechanism analysis, providing strong support for modern agricultural biology research.
References
- Liu, J., Chen, S., Liu, M et al. (2022). Full-Length Transcriptome Sequencing Reveals Alternative Splicing and lncRNA Regulation during Nodule Development in Glycine max. International journal of molecular sciences, 23(13), 7371. https://doi.org/10.3390/ijms23137371
- Schaarschmidt, S., Fischer, A., Lawas, L. M. F., Alam, R et al. (2020). Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L. International journal of molecular sciences, 21(21), 8148. https://doi.org/10.3390/ijms21218148
- Wang, W., Wu, L., Shi, Y., Yin, Q et al. (2024). Integrated Full-Length Transcriptomics and Metabolomics Reveal Glycosyltransferase Involved in the Biosynthesis of Flavonol Glycosides in Laportea bulbifera. Journal of agricultural and food chemistry, 72(14), 8269–8283. https://doi.org/10.1021/acs.jafc.4c00488
- Zheng, S. Y., Pan, L. X., Cheng, F. P., Jin, M. J., & Wang, Z. L. (2023). A Global Survey of the Full-Length Transcriptome of Apis mellifera by Single-Molecule Long-Read Sequencing. International journal of molecular sciences, 24(6), 5827. https://doi.org/10.3390/ijms24065827
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment