We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
PacBio HiFi Sequencing
At a glance:
- What Is HiFi Sequencing?
- What Are HiFi Reads?
- Benefits of HiFi Sequencing
- How HiFi Sequencing Works?
- Applications of HiFi Sequencing
PacBio sequencing technology has evolved to a different type of long read, known as highly accurate long reads, or HiFi reads. PacBio, the only sequencing technology to offer HiFi reads, is 99.9% accurate, comparable to short reads and Sanger sequencing. With HiFi reads, you no longer need to sacrifice long read lengths for high-precision sequencing to solve the toughest biological problems, such as single nucleotide and structural variant detection, genome assembly, assembly of difficult polyploid or highly repetitive genomes, and assembly of metagenomes.
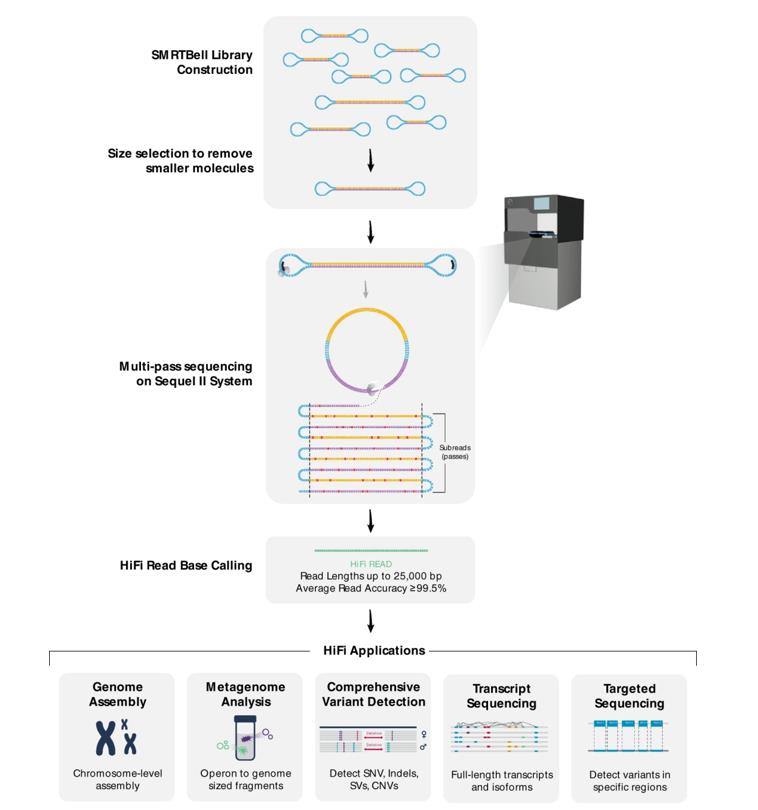 Flowchart of HiFi sequence read generation and downstream applications. (Hon et al., 2020)
Flowchart of HiFi sequence read generation and downstream applications. (Hon et al., 2020)
What Is HiFi Sequencing?
HiFi sequencing is the core technology underpinning PacBio's long-read sequencing platform, which seamlessly combines the benefits of short-read sequencing and traditional long-read sequencing. HiFi sequencing allow you to accurately detect all types of variants, from single nucleotides to structural variants, with high precision, recall, and in-phase haplotypes, even in difficult-to-sequence regions of the genome. Its availability raises the bar for precision and accuracy in genome sequencing, enabling scientists to explore deeper and with greater confidence.
What Are HiFi Reads?
HiFi reads are generated on the PacBio Long Read System using Cyclic Consistent Sequencing (CCS) mode. HiFi reads produce highly accurate long read sequencing datasets, with an average read length of 10-25 kb and an accuracy of greater than 99.5%. HiFi reads can be used in a wide variety of SMRT sequencing applications, including whole genome sequencing from de novo assemblies, comprehensive mutation detection, epigenetic characterization, RNA sequencing, and other genome sequencing applications.
Benefits of HiFi Sequencing
Accuracy
Sequencing without systematic errors achieves consensus accuracy of >99.999% HiFi sequencing provides the information needed to confidently call and detect all variants with superior accuracy, low sequencing background bias, and accurate read mapping.
Long Reads
Traditional long-read technologies often require a trade-off between read length and accuracy. However, with HiFi sequencing, researchers can benefit from long reads, often spanning tens of bases, without compromising data integrity.
Uniform Coverage
One of the distinguishing features of HiFi sequencing is its ability to provide even coverage. Unlike other methods, HiFi sequencing does not show bias based on GC content, making it possible to sequence challenging genomic regions, from AT-rich sequences to highly repetitive GC-rich sequences.
Single-molecule Resolution
The technology captures sequence data directly from natural DNA or RNA molecules, ensuring the generation of highly accurate long reads with stunning single-molecule precision.
Direct Epigenetic Testing
HiFi Sequencing allows direct detection of base modifications during sequencing due to the lack of a PCR amplification step. This enables the capture of both sequence and epigenetic data in a unified experiment.
How HiFi Sequencing Works?
The HiFi sequencing process is characterized by multiple cycles of consistent sequencing of long individual molecules, typically up to ~25kb in length. Starting with a high-quality DNA sample, scientists prepare an SMRTbell library. Once the library is ready, it is sequenced on the PacBio Sequel IIe system. After the data is generated, the raw reads are subjected to a cycle-consistent sequencing process to produce the final HiFi reads.
Sequencing begins when a looped fragment of sample DNA suspended in solution slips through a nanofluidic chip called an SMRT (Single Molecule Real Time) cell. The design of this SMRT cell is fascinating. It is intricately packed with millions of cylindrical grooves, or zero-mode waveguides (ZMWs), each just a few nanometers wide.
When the sample is slid, the DNA fragments find their place at the bottom of these ZMWs. Free nucleotides are then added to the mixture. There is also a DNA polymerase, which is attached in the early stages of library preparation and begins to function. It begins to replicate the DNA sample. Each time a nucleotide is mixed into this newly formed strand, a tiny amount of light is released. When this light is detected, the system recognizes which DNA base (adenine, thymine, cytosine, or guanine) it contains based on its specific pattern.
Multiple copies of each DNA fragment are generated within the ZMW. This sequencing of duplicates gives HiFi amazing accuracy. Cross-referencing of each DNA copy, a method known as circular coherence sequencing (CCS), helps to identify the exact sequence of the sample.
In addition, the Sequel IIe system and other state-of-the-art PacBio sequencing platforms record the rate at which the polymerase dopes each base. PacBio's proprietary SMRT Link software then parses that rate to determine whether the base is methylated or not-a key indicator for in-depth epigenetic studies.
Applications of HiFi Sequencing
Haplotype Profiling
Deciphering the genetic code to reveal the basis of certain traits, especially in crops or human genetic diseases, requires distinguishing each chromosome copy or haplotype. This distinction is known as phasing and is helpful. With HiFi sequencing, the overall process of phasing is simplified and refined. This eliminates the traditional reliance on trio or population-based techniques, which often require a significant investment of time and resources.
For example, when researchers delved into the genomics of spinal muscular atrophy (SMA), they used HiFi sequencing. The result was the identification of two SMN1 haplotypes in an African population, providing transformative insights into carrier risk indicators.
Mutation Detection and Genome Assembly
PacBio HiFi sequencing has an unrivaled ability to span a wide range of genomic regions, making it an indispensable tool for detecting genetic variation. Regions with exceptionally large insertion-deletion events or regions with highly repetitive sequences, traditionally considered challenging, can be accessed with HiFi's long and precise reads.
In addition, HiFi sequencing is a pioneer in the assembly of genomes of multiple life forms. The precision and length of the data it provides ensures overlap between reads, even in regions of high homology. This overlap ensures that assembly software can reconstruct the genome in a way that reduces errors and ambiguity. Notably, this strength is demonstrated when the T2T Consortium utilizes HiFi sequencing to complete the human genome assembly in 2022.
With HiFi sequencing's ability to analyze samples directly, without an amplification phase, researchers can access critical base modification data. This opens up a wide range of possibilities for research that strives to understand genetic changes in gene expression. Additionally, when used in conjunction with other HiFi applications, this methylation data allows researchers to identify and detect epigenetic effects in a thoroughly phased genomic environment.
Reference
- Hon, Ting, et al. "Highly accurate long-read HiFi sequencing data for five complex genomes." Scientific data. 7.1 (2020): 399.
Related Services
PacBio SMRT Sequencing Technology
PacBio Pre-Made Library Sequencing
Epigenetics and Methylation Analysis Using Long-Read Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment