

We are dedicated to providing outstanding customer service and being reachable at all times.
PacBio HiFi Metagenomics
At a glance:
- Overview
- Advantages of PacBio HiFi Metagenome Sequencing
- Workflow of PacBio HiFi Metagenome Sequencing
- Hifiasm-meta Boosts the Power of HiFi Metagenomes
Overview
Metagenome sequencing has historically been a challenging endeavor. The inherent complexity of microbial communities means that any sequencing technology must provide both breadth (to capture entire communities) and depth (to understand individual species). This balance is critical for reconstructing the accurate genome of each member, especially those that are less abundant or uncultured.
PacBio HiFi metagenomics is becoming the method of choice for microbiome research. Unlike other sequencing methods, HiFi provides high-precision long-read sequencing. Each HiFi read captures contiguous long DNA fragments with an accuracy comparable to the short reads traditionally used in metagenomics. This approach is capable of spanning and accurately sequencing entire genes, operons, and even complete microbial genomes.
HiFi metagenomic sequencing has been applied to the study of human gut samples. By focusing only on circular contigs, the researchers assembled 102 complete metagenome amssembled genomes (MAGs) from just five human stool samples. In contrast, an earlier attempt using Oxford Nanopore sequencing obtained only 20 cMAGs from 13 samples.
Accurate, flexible HiFi solutions for metagenomics and microbiome sequencing enable researchers to study microbes and microbial communities at high resolution without the need for culturing. The superior accuracy and long reads of HiFi sequencing give scientists the flexibility and high resolution they need.
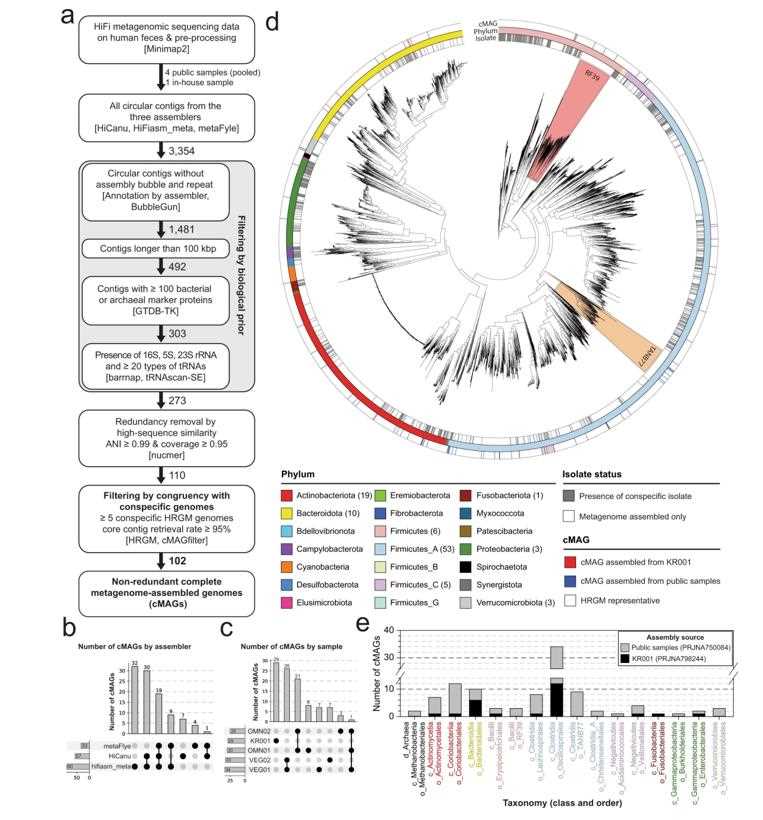 Assembly of 102 complete metagenome-assembled genomes (cMAGs) from human fecal high-accuracy long-read (HiFi) metagenomic sequencing samples. (Kim et al., 2022)
Assembly of 102 complete metagenome-assembled genomes (cMAGs) from human fecal high-accuracy long-read (HiFi) metagenomic sequencing samples. (Kim et al., 2022)
Advantages of PacBio HiFi Metagenome Sequencing
Species and Strain-level Resolution
One of the major challenges in metagenomic research is obtaining species and strain level resolution. Traditional short-read sequencing methods often fail to provide a comprehensive understanding of microbial communities. With its exceptional accuracy, HiFi sequencing provides species-level resolution, allowing researchers to pinpoint the microbial composition of a sample.
For example, full-length 16S rRNA sequencing provided by PacBio HiFi Sequencing provides insight into taxonomic classifications. This detailed sequencing can clearly distinguish strains of similar species, such as E. coli O157:H7, providing researchers with a clear understanding of microbial ecosystems.
Comprehensive Metagenomic Analysis
PacBio HiFi Sequencing goes beyond mere taxonomic classification. Capable of capturing 6-8 full-length genes per HiFi read, it provides an efficient and cost-effective method for metagenomic analysis. This capability ensures that researchers can not only identify microbial entities but also gain insight into their functional role in the community.
For example, with such an in-depth analysis, it became feasible to examine the functional capacity of the microbial community responsible for digesting plastic waste. By understanding collective functional roles, researchers can potentially enhance and simplify processes such as plastic degradation.
High-Quality Metagenome Assembled Genomes (MAGs)
A prominent feature of PacBio HiFi Sequencing is the ability to produce high-quality MAGs. For example, on a Sequel II or IIe system, up to 45 MAGs can be generated for multiple human stool samples. The ability to obtain such extensive metagenomically assembled genomes has revolutionized the scope of metagenomic research, allowing access to detailed genomic blueprints of previously uncharacterized microbial entities.
Enhanced Insights Using Epigenomic Data
The fusion of metagenomics and epigenomics demonstrates the versatility of PacBio HiFi sequencing. Researchers can now use epigenomic data to associate contigs and plasmids of closely related strains. This association adds another layer of precision to metagenomic studies, ensuring that even the tiniest genomic variation does not go unnoticed.
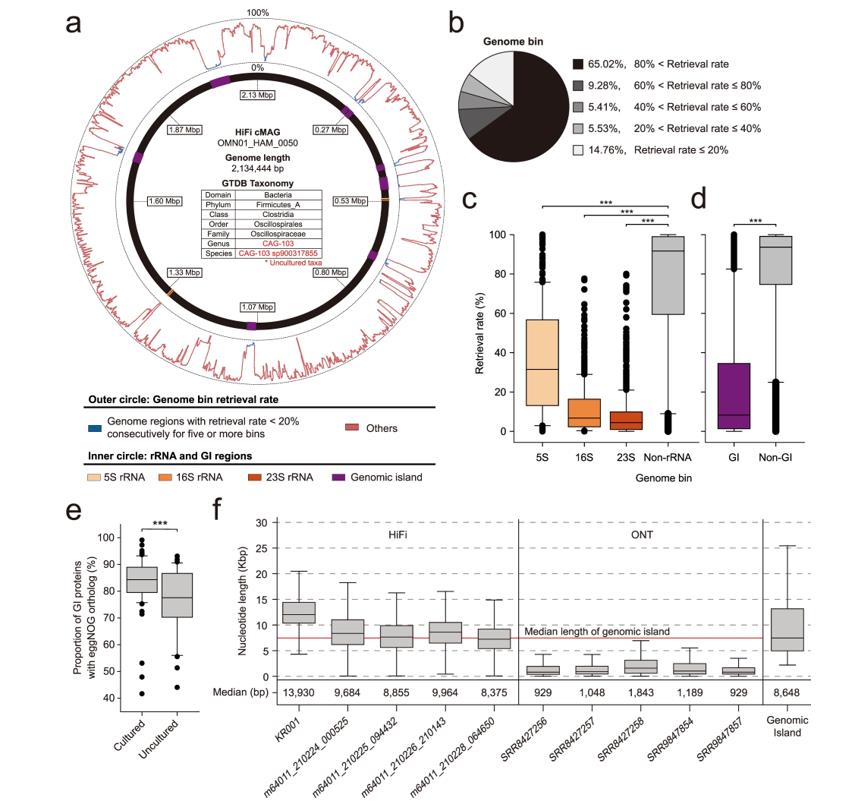 HiFi-assembled cMAGs retrieve hard-to-assemble regions by short-read assembly. (Kim et al., 2022)
HiFi-assembled cMAGs retrieve hard-to-assemble regions by short-read assembly. (Kim et al., 2022)
Workflow of PacBio HiFi Metagenome Sequencing
(1) Sample preparation: Start by isolating high-quality DNA from the microbial community of interest.
(2) Library construction: Prepare DNA for sequencing using PacBio's protocol. This involves creating SMRTbell libraries optimized for HiFi sequencing.
(3) Sequencing: Prepared libraries were sequenced using PacBio's Sequel system. The system generates HiFi reads by repeatedly sequencing each SMRTbell library molecule.
(4) Assembly: With HiFi reads, specialized assembly programs such as hifiasm-meta or metaFlye are used to reconstruct the genome present in the sample.
(5) Analysis: Dive deeper into assembled genomes. This may involve annotating genomes, understanding their metabolic potential, or comparing them to known microbial genomes to determine taxonomy.
Hifiasm-meta Boosts the Power of HiFi Metagenomes
An evolutionary leap from PacBio HiFi sequencing combined with the hifiasm-meta tool. Hifiasm-meta not only utilizes long, accurate HiFi reads, but reconstructs them with unparalleled accuracy. In empirical tests, it consistently reconstructed tens to hundreds of complete circular bacterial genomes in each dataset, some even exceeding the 1Mb threshold. This is an achievement unmatched by other metagenomic assemblers.
With hifiasm-meta, PacBio reaffirms its commitment to the in-depth study of microbial communities. The ability to generate a nearly complete metagenomic picture brings many advantages:
- Enhanced taxonomic resolution: Hifiasm-meta enables researchers to discriminate microbial data down to the species or even strain level. This level of detail is invaluable when trying to understand the roles and interactions of specific microbial entities in a community.
- Functional insights: Beyond understanding the members of a microbiome, the real gold lies in understanding what they do. Hifiasm-meta, when combined with PacBio HiFi sequencing, can provide deep functional insights and provide a clearer picture of metabolic pathways and interactions.
- Study cost-effectiveness: HiFi metagenomic downsampling experiments show that a sample run on 4 SMRT cells can obtain a data width similar to 48 multiplex samples. Not only does this mean cost savings, but it also ensures access to state-of-the-art insights without exponentially growing resources.
Reference
- Kim, Chan Yeong, Junyeong Ma, and Insuk Lee. "HiFi metagenomic sequencing enables assembly of accurate and complete genomes from human gut microbiota." Nature communications. 13.1 (2022): 6367.
Related Services
PacBio SMRT Sequencing Technology
Long-Read Metagenomics Sequencing
Full-Length 16S/18S/ITS Amplicon Sequencing
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment