We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Fusion Transcript Sequencing
At a glance:
- Overview
- Traditional Short-read RNA-Seq for Detection of Fusion Transcripts
- Long-read RNA-Seq: New Possibilities for Fusion Transcript Discovery
- Workflow for Long-Read RNA-Seq Detection of Fusion Transcripts
- Bioinformatic Approaches for Fusion Detection in Long-read Data
Overview
Fusion genes are caused by chromosomal aberrations such as translocations, duplications, inversions or small interstitial deletions. At the transcript level, fusion genes may not only reflect underlying genomic rearrangements, but may also arise as a result of aberrant transcription or trans-splicing events. Fusion genes are a major cause of cancer, accounting for approximately 20% of human cancer incidence. However, the prevalence of fusion genes varies widely across cancers and many fusion genes are specific to certain cancer subtypes. Therefore, rapid and accurate identification of fusion genes can characterize and stratify a cancer diagnosis and inform subsequent treatment.
Fluorescence in situ hybridization (FISH) and quantitative real-time polymerase chain reaction (RT-PCR) methods are primarily used for fusion gene diagnosis. Although highly sensitive, these methods typically test for the presence of only a single fusion gene, often resulting in a lengthy, iterative and costly diagnostic process. In addition, these methods are unable to identify novel fusion gene partners or resolve complex structural rearrangements.
With the avant-garde advent of RNA sequencing (RNA-seq) and Nanopore sequencing technologies, the landscape of fusion gene detection has witnessed a paradigmatic shift. These technologies elucidate full-length transcript sequencing with unprecedented read lengths, proffering profound insights into gene fusion dynamics.
Traditional Short-read RNA-Seq for Detection of Fusion Transcripts
RNA-Seq has long been a traditional method for transcriptome research, capable of identifying fusion transcripts. The sensitivity and specificity of the method depends on the sequencing depth, read length and quality, as well as the bioinformatics methods and parameters used. However, traditional short read length sequencing faces several challenges:
- Fragmentation and assembly: Sequencing involves fragmenting cDNA libraries and reading them in short sequences (~50-100 bases). After sequencing, computational assembly is required to infer the complete transcript sequence. This fragmentation often leads to misassembly, especially in the case of detecting split-sequence (SAS) fusions.
- Complex genomic regions: Short read lengths make it difficult to capture complex genomic rearrangements, repeat-rich regions, or full-length transcripts. Such limitations require complex computational analyses to infer full-length transcript sequences, leading to the potential omission of biologically significant variants.
To circumvent these challenges, bioinformaticians have developed two main strategies: a mapping-first approach, which identifies inconsistent reads indicative of genomic rearrangements; and an assembly-first approach, in which reads are assembled into longer transcript sequences to discover fusion transcripts.
Synthetic long read length (SLR) sequencing has been introduced as an alternative, aiming to combine the advantages of the thoroughness of long read length sequencing with the cost-effectiveness and accuracy of short read length sequencing. Here, short reads sharing the same barcode are compiled to construct longer reads.SLR-seq has been successfully used to identify large-scale isoform redistribution and several previously unknown fusion isoforms in benign colonic mucosa, primary colon cancer and metastatic colon cancer. While SLR provides deeper insights, it still relies on short reads as the basic assembly unit, limiting its efficacy to specific regions and capturing large numbers of repeats.
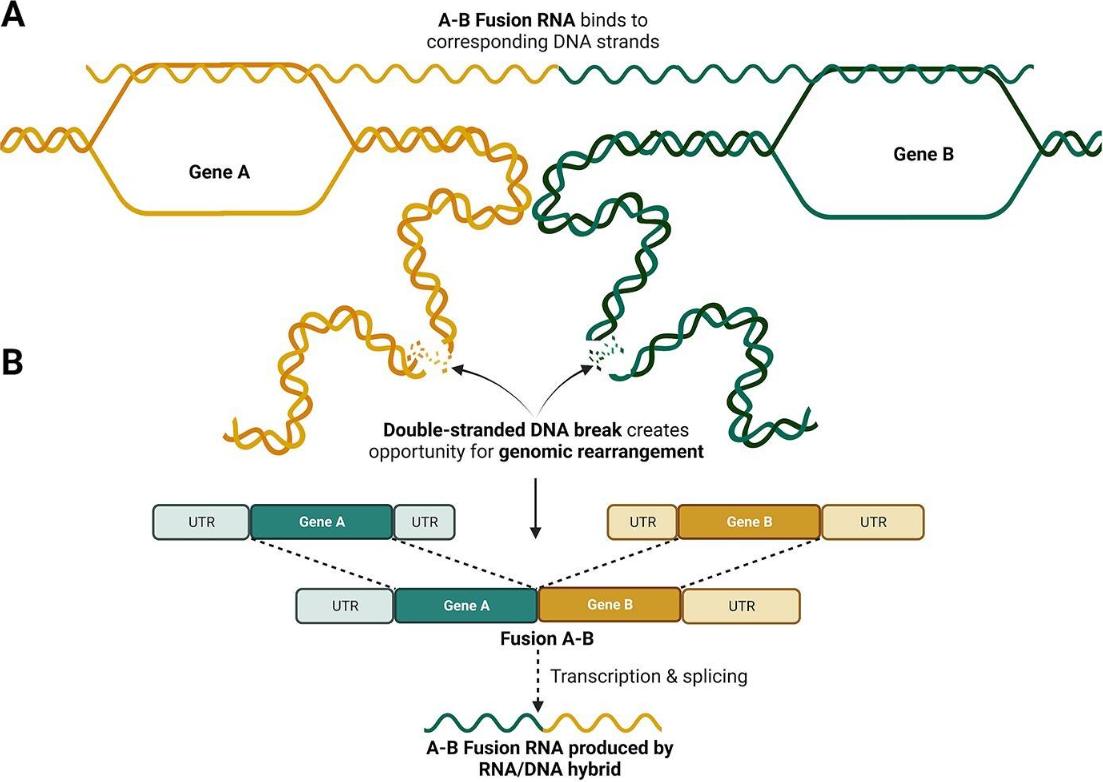 Theoretical model of RNA-mediated gene fusions. (Dorney R et al., 2023)
Theoretical model of RNA-mediated gene fusions. (Dorney R et al., 2023)
Long-read RNA-Seq: New Possibilities for Fusion Transcript Discovery
Advances in long-read sequencing technologies such as PacBio and Oxford Nanopore Technology (ONT) have enabled the generation of read lengths of tens of kilobases in length at relatively low cost, providing a more comprehensive view of transcripts. Although longer read length sequencing is more expensive than short read length sequencing, long read lengths can produce more accurate fusion predictions with the following advantages:
Full-length sequencing: the ability to span the full length of a transcript improves localization accuracy and promotes unambiguous identification of fusion transcripts.
Complex isoform detection: complex multi-exon isoforms, large transcripts, and complex fusion types (e.g., double-hop and bridging fusions) can be resolved without computational inference.
Oxford Nanopore's MinION system stands out for its real-time data generation capabilities. Using this system, assays have been developed that can detect oncogenic gene fusions in a very short period of time. By modifying the anchored multiplex PCR method for library creation, fusions such as BCR-ABL1 can be identified within minutes of sequencing initiation.
using the concepts written previously, rewrite this article with a high degree of complexity and specificity:
Workflow for Long-Read RNA-Seq Detection of Fusion Transcripts
(1) Fusion Transcript Enrichment
To ensure accurate detection of fusion transcripts, the first step is enrichment, using two main techniques:
Sequence-specific RT-PCR:
When dealing with a known complete transcript, a sequence-specific RT-PCR approach is adopted. This is a highly targeted amplification technique tailored to ensure the detection and amplification of the exact fusion transcript under scrutiny. Its precision stems from its ability to focus solely on the fusion transcript of known sequence composition.
Semi-specific RT-PCR:
In instances where the knowledge encompasses only one end of the fusion transcript, semi-specific RT-PCR becomes the method of choice. This technique amplifies transcripts by leveraging known fragment sequences, thus presenting a pathway to unearth previously undetected fusion events.
(2) Sequencing
After the enrichment phase, the next procedural step is the meticulous preparation of the library. At this juncture, the PCR-cDNA Sequencing Kit emerges as a pivotal tool. It presents a streamlined mechanism to efficiently transmute enriched RNA samples into sequencing-ready libraries, ensuring the fidelity of the transcript representation.
(3) MinION and Flongle Flow Cells: determining the size and depth of sequencing
MinION:
The MinION's compact footprint belies its capabilities. It's a quintessential device for real-time sequencing applications, boasting versatility that allows its deployment in standard laboratory conditions or even in more challenging remote research environments.
GridION:
Engineered with multifaceted projects in mind, the GridION houses up to five individually addressable flow-through tanks. This intricate design strikes a harmonious chord between scalability-catering to expansive sequencing needs-and the granularity of a detailed molecular analysis.
(4) Sequencing Equipment: Flexibility in the sequencing process can be increased by choosing equipment
MinION:
The MinION's compact footprint belies its capabilities. It's a quintessential device for real-time sequencing applications, boasting versatility that allows its deployment in standard laboratory conditions or even in more challenging remote research environments.
GridION:
Engineered with multifaceted projects in mind, the GridION houses up to five individually addressable flow-through tanks. This intricate design strikes a harmonious chord between scalability – catering to expansive sequencing needs – and the granularity of a detailed molecular analysis.
(5) Data Analysis
Post-sequencing, the generated data necessitates rigorous computational and statistical scrutiny. This phase involves the application of robust bioinformatics tools and algorithms to ascertain the veracity of detected fusion transcripts, quantifying their abundance, and contextualizing their potential biological implications.
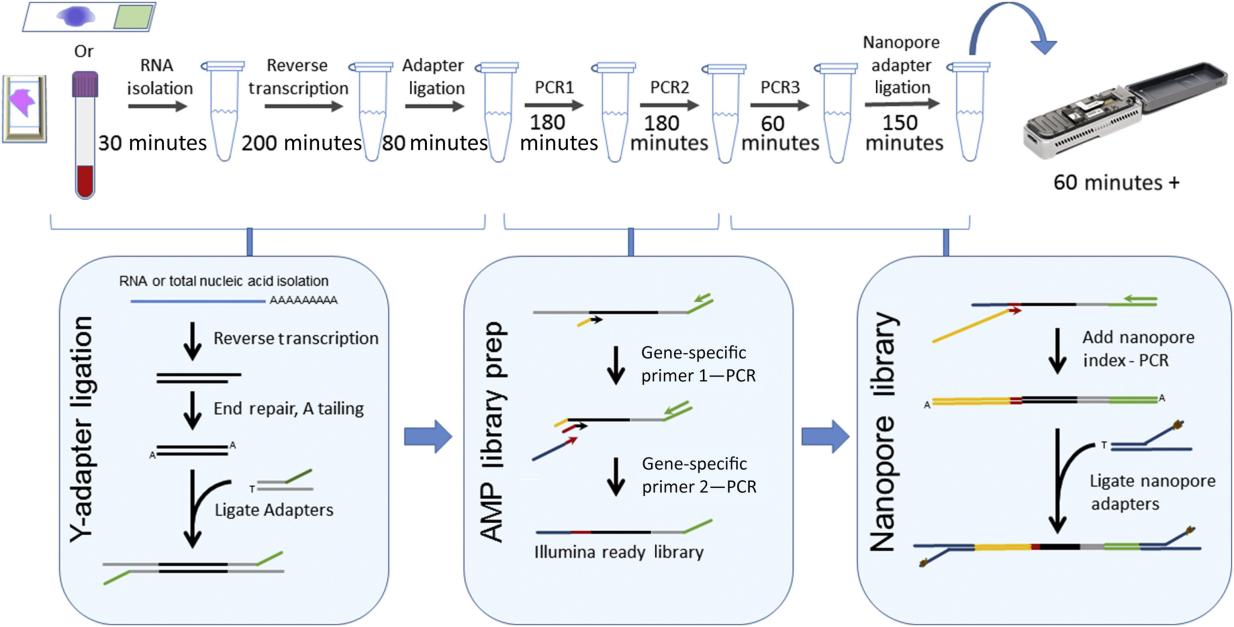 Multistep anchored multiplex PCR (AMP)-based library preparation for MinION sequencing and turnaround time. (Jeck W R et al., 2019)
Multistep anchored multiplex PCR (AMP)-based library preparation for MinION sequencing and turnaround time. (Jeck W R et al., 2019)
Bioinformatic Approaches for Fusion Detection in Long-read Data
LongGF: A Pioneering Approach
One of the first tools tailored for long-read fusion detection was LongGF. Utilizing Minimap2 for genome alignment, it offers a prioritized list of potential gene fusions. A unique feature of LongGF is its ability to filter out overlapping genes and alignments, clustering reads for a more concise fusion detection. However, its reliance on predefined genomic coordinates can be a double-edged sword. While it provides specificity, it may miss out on detecting fusions involving uncharacterized genes or exons. Moreover, the sequence similarity between homologous genes might pose challenges in determining the origin of fusion partners.
Genion: Stringent Fusion Filtering
To address the potential pitfalls of false positives, Genion emerged with more stringent thresholds. Using desalt for genome alignment, this tool applies filters not to individual reads but to entire read clusters. Such an approach offers powerful filtering, presenting cleaner candidates for analysis. Genion's strength lies in its ability to discern between genuine fusion events and mapping errors or genomic variants. However, its rigorous filtering might sometimes be over-cautious, leading to potential overlooks of valid fusion events.
AERON: Translating Reads to Transcriptomes
A standout tool, AERON, opted for a different alignment strategy. Using GraphAligner, AERON aligns reads directly to a reference transcriptome, bypassing the genome. This approach allows for the quantification of transcripts, translating read counts into Transcripts Per Million values. While innovative, aligning to the transcriptome introduces challenges, especially when dealing with highly similar short-length transcripts.
JAFFAL: Dual Alignment for Precision
Building upon the foundation of its predecessor, the short-read fusion caller JAFFA, JAFFAL employs a two-pronged alignment strategy. Using Minimap2, reads are first aligned to a reference transcriptome, followed by a secondary alignment to a reference genome for reads indicating potential fusions. This dual-alignment strategy not only minimizes false positives but also streamlines computational demands, given that only a subset of the reads undergo genome alignment.
References
- Dorney, Ryley, et al. "Recent advances in cancer fusion transcript detection." Briefings in Bioinformatics 24.1 (2023): bbac519.
- Jeck, William R., et al. "A nanopore sequencing–based assay for rapid detection of gene fusions." The Journal of Molecular Diagnostics 21.1 (2019): 58-69.
Related Services
Oxford Nanopore Sequencing
Pacbio RNA Sequencing
Nanopore RNA Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment