We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Structural Variation Analysis
At a glance:
- Overview
- Definition and Types of Structural Variants
- Generation of Structural Variants
- Methods for Characterizing Structural Variants
- DeBreak, combiSV, and cuteSV: New Software Tools for Structural Variants
Overview
Structural variants (SVs) represent significant alterations in DNA beyond the length of typical single nucleotide polymorphisms. As the largest variation in the human genome, SVs are closely associated with human diseases (e.g., hereditary disorders and cancer), evolution (e.g., gene loss and transposon activity), gene regulation (e.g., rearrangement of transcription factors), and other phenotypes (e.g., mating and intrinsic reproductive isolation). Given their complex nature and a huge impact on the genomic landscape, efficient and accurate SV detection methods are essential.
Efforts have been made to develop short-read based SV calling methods. Most of them use methods such as read depth, incongruent read pairs, split read alignment, local assembly, or combinations thereof, and they have an important role in large-scale genomics studies such as the Thousand Genomes Project. However, relatively low read lengths limit these tools from achieving sensitive SV detection and also suffer from false positives.
With the rapid development of long-read sequencing technologies, such as the Pacific Bioscience and Oxford Nanopore Technology platforms, which can span SVs end-to-end, an opportunity exists to detect SVs more comprehensively at higher resolution. However, new computational methods are needed to deal well with the high sequencing error rates (typically 5-20%) and large lengths (on average more than 10kbp) of reads.
Based on long-read sequencing platforms, Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), CD Genomics can help customers comprehensively detect SVs at a higher resolution.
Definition and Types of Structural Variants
SVs are large genomic alterations, usually defined as deletions (DEL), insertions (INS), duplications (DUP), inversions (INV), and translocations (TRA) of at least 50 bp in size. SVs are considered separately from small variants, which include single nucleotide variants (SNVs) and short insertions and deletions (indel) because they are usually formed by different mechanisms. sVs can be equilibrium, with There is no loss or gain of genetic material, such as INV and TRA. unbalanced DELs and DUPs are also known as copy number variants (CNVs). SVs are largely responsible for the diversity and evolution of the human genome at both the individual and population levels. SVs have a greater impact on gene function and phenotypic changes than SNVs and indel. As a result, SVs are associated with many human diseases, including neurodevelopmental disorders and cancer.
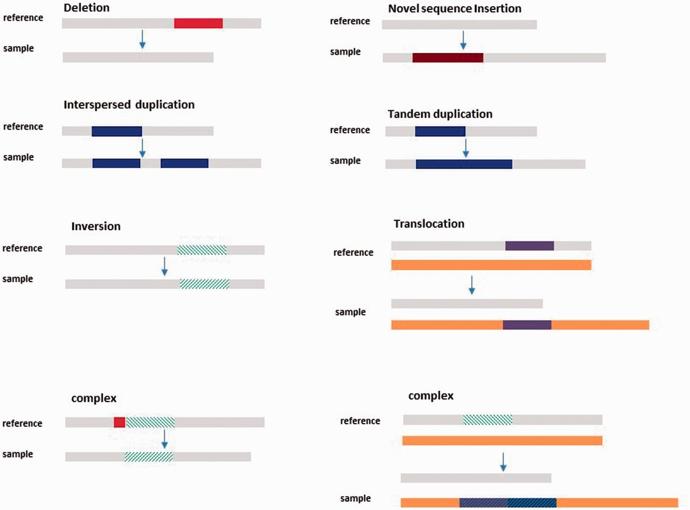 Types of structural variants. (Escaramís et al., 2015)
Types of structural variants. (Escaramís et al., 2015)
Generation of Structural Variants
Multiple molecular mechanisms drive SV generation. These can arise through meiosis and mitosis. Key mechanisms include:
- Non-allelic homologous recombination (NAHR): a recombination event that occurs as a result of mismatches in closely related sequences during replication.
- Non-homologous end joining (NHEJ): a mechanism for the repair of mammalian double-strand breaks.
- Microhomology-mediated end joining (MMEJ): a variant of NHEJ that is more error-prone.
- Fork stopping and template switching (FoSTeS) and microhomology-mediated break-induced replication (MMBIR): mechanisms involved in the generation of complex genomic rearrangements.
Methods for Characterizing Structural Variants
Microarrays
Before the rise of high-throughput sequencing, microarrays were the platform of choice for detecting SVs. Microarrays rely on the hybridization of labeled DNA fragments to an array of DNA probes on a solid surface.
Advantages:
- High throughput and cost-effectiveness for large-scale studies.
- Capable of detecting variation across the genome.
Limitations:
- Limited to detecting copy number variation.
- Cannot recognize SV breakpoints or precise locations.
High-Throughput Sequencing-Based Approaches
With the advent of Next Generation Sequencing (NGS) technology, a new era of SV detection has been ushered in. There are several ways to utilize the power of NGS:
| Methods | Description | Advantages | Limitations |
| Raired Read Analysis | The distance between reads at both ends is analyzed. If the observed distance between reads deviates significantly from the expected distance, it may indicate the presence of SV. | Can detect deletions, insertions, and inversions. Provides an accurate estimate of SV size. |
Requires high coverage for accurate detection. |
| Split Read Analysis | This method identifies SVs by finding reads that span a breakpoint. The portion of the read aligned to both sides of the breakpoint can be used to infer the type and size of the variant. | Provides accurate identification of breakpoints. Smaller SVs can be detected. |
Requires high read length to ensure accuracy. |
| Assembly-based Analysis | In this method, ab initio assembly of the genome is performed. The assembled sequence is then compared to a reference genome to identify structural variants. | Provides comprehensive SV detection without relying on a reference genome. Can detect complex SVs. |
Computationally intensive and time-consuming. |
| Depth of Coverage Analysis | The depth of sequencing reads across the genome is measured. Regions where the read depth is significantly higher or lower than expected indicate potential duplications or deletions, respectively. | Ideal for detecting copy number variants. Less dependent on read length. |
Cannot detect balanced SVs such as inversions. |
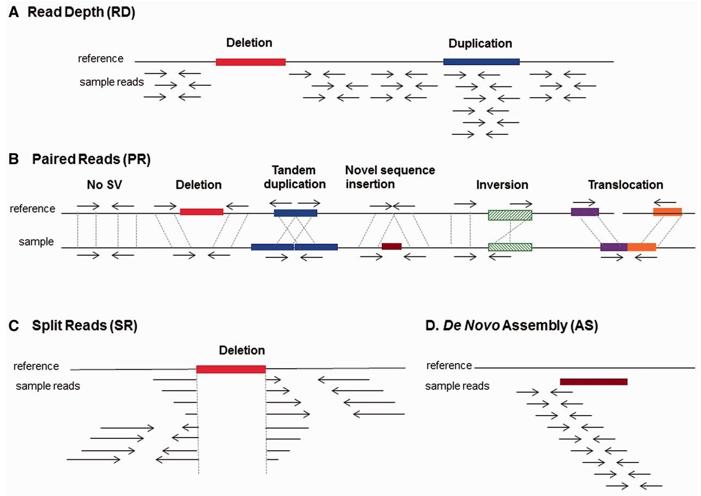 Strategies for structural variant detection. (Escaramís, et al., 2015)
Strategies for structural variant detection. (Escaramís, et al., 2015)
The advent of long-read sequencing technologies has dramatically reshaped the landscape of genomic research. Unlike NGS, which typically provides shorter reads, long-read sequencing produces much longer read lengths, often spanning hundreds of bases. This considerable length advantage provides a comprehensive view of complex regions, enabling the detection of larger SVs that may be missed or fragmented by shorter reads.
In addition, long-read sequencing has a unique advantage in analyzing SVs located within repetitive or GC-rich regions, such as repeat extensions, which often pose a challenge to other sequencing methods. By being able to span the entire SV end-to-end, long-read long sequencing provides unrivaled resolution, revealing the SV and its potential epigenetic impact.
DeBreak, combiSV, and cuteSV: New Software Tools for Structural Variants
DeBreak: precise structural variant discovery
DeBreak emerges at the forefront of the computational realm as an intricate engine formulated to achieve a broad yet nuanced detection of structural variants (SVs). Its prowess is underscored by a bipartite methodology tailored for SV elucidation. When addressing SVs manifesting within reads, DeBreak deploys an intricate scan on the reads for an in-depth comparative assessment. It subsequently employs a density-oriented clustering algorithm, designed to aggregate these signals. The final refinement phase harnesses the capabilities of the partial order alignment (POA) algorithm, a cutting-edge computational method, that aids in the meticulous identification of SV breakpoints right to the atomic resolution of a single base pair. For SVs of a magnitude that overshadows the conventional read length, DeBreak's integrative architecture incorporates local de novo assembly, thereby reconstructing sequences encapsulating the SV. The culmination of these processes results in DeBreak's standing as an essential instrument in the domain of structural variation analytics.
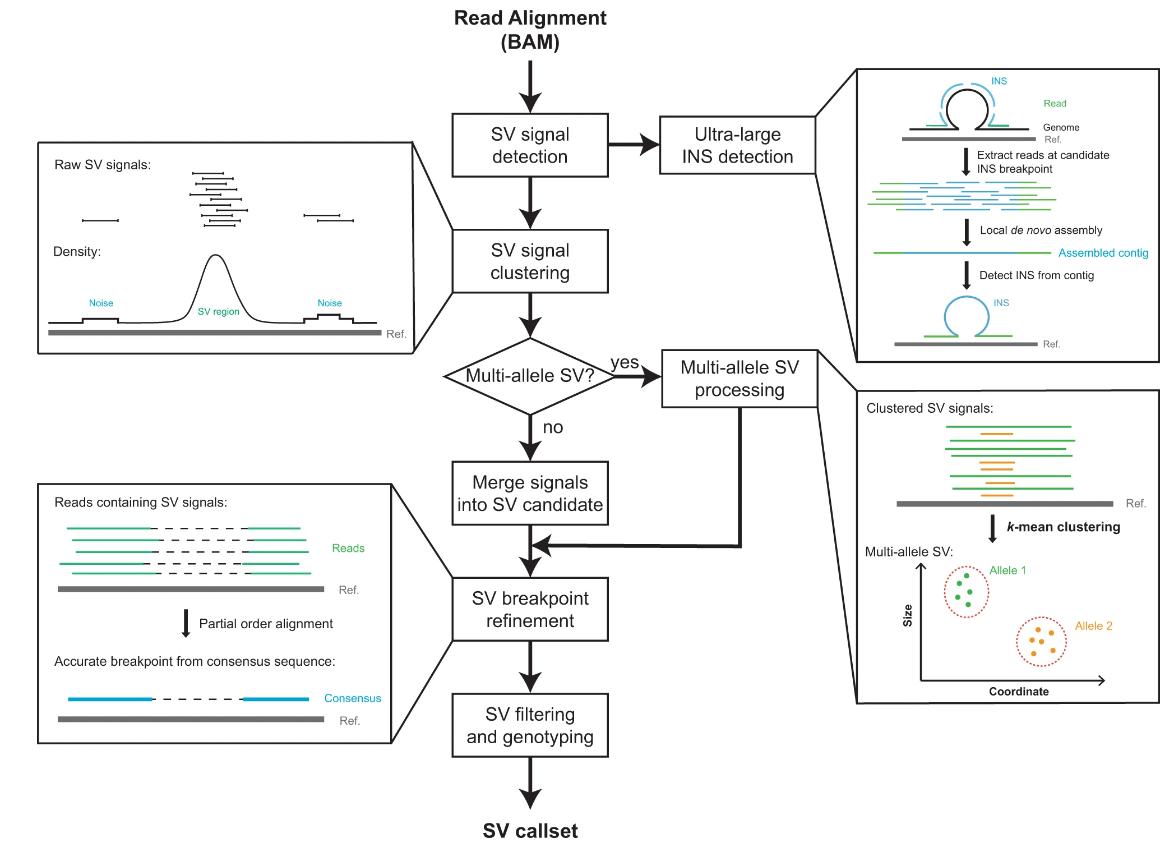 The major steps of DeBreak SV discovery include SV signal detection, signal clustering, breakpoint refinement, and filtering and genotyping. (Chen et al., 2023)
The major steps of DeBreak SV discovery include SV signal detection, signal clustering, breakpoint refinement, and filtering and genotyping. (Chen et al., 2023)
CombiSV: integrating structural variation results for enhanced detection
Expanding upon the foundational principles in contemporary SV research, combiSV pioneers an innovative trajectory by fusing outputs from an array of structural variant calling instruments. This integration is strategized to proffer a SV dataset that is both exhaustive and precision-driven. By seamlessly knitting together the inherent capabilities of each SV caller, combiSV not only heightens recall but also amplifies precision metrics. A testament to its prowess is the compelling performance when analyzed alongside the Genome in a Bottle (GIAB) consortium's gold-standard benchmark set, which further accentuates combiSV's formidable efficacy.
CuteSV: efficient and scalable SV detection
Distinctively positioned, CuteSV revolutionizes the domain with its comparison-centric SV detection mechanism, meticulously curated for long read-length sequencing datasets. At the core of CuteSV's architecture is its capability to aggregate features spanning diverse SV genres. This then cascades into a sharp SV detection modus operandi, leveraging both clustering and refinement methodologies. While the current gamut of tools often oscillate between sensitivity, scalability, and specificity, particularly concerning distinctive sequencing platforms, CuteSV emerges as an exemplar, boasting cross-platform adaptability, swifter computational throughput, and an unparalleled sensitivity spectrum, a facet that is pronounced even when confronted with datasets characterized by reduced coverage.
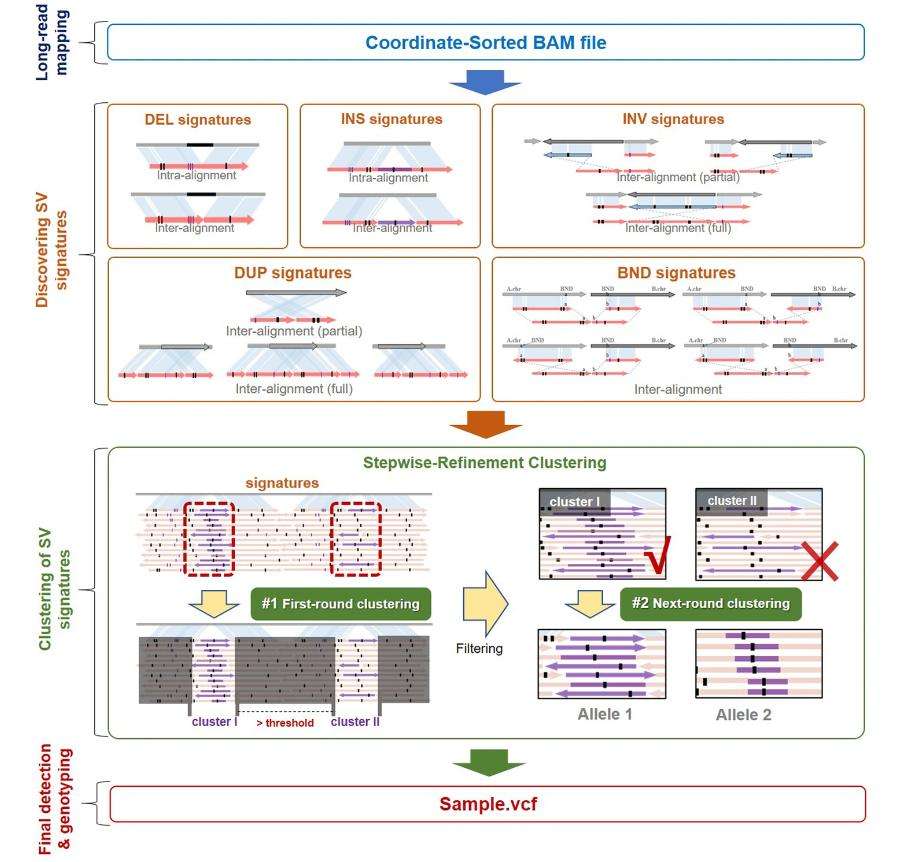 Schematic illustration of the cuteSV approach. (Jiang et al., 2020)
Schematic illustration of the cuteSV approach. (Jiang et al., 2020)
References
- Escaramís, Geòrgia, Elisa Docampo, and Raquel Rabionet. "A decade of structural variants: description, history and methods to detect structural variation." Briefings in functional genomics 14.5 (2015): 305-314.
- Chen, Yu, et al. "Deciphering the exact breakpoints of structural variations using long sequencing reads with DeBreak." Nature Communications 14.1 (2023): 283.
- Dierckxsens, Nicolas, et al. "A benchmark of structural variation detection by long reads through a realistic simulated model." Genome Biology 22.1 (2021): 1-16.
- Jiang, Tao, et al. "Long-read-based human genomic structural variation detection with cuteSV." Genome biology 21.1 (2020): 1-24.
Related Services
PacBio SMRT Sequencing
Oxford Nanopore Sequencing
Variant Calling
Human Genome Structural Variation Detection
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment