
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
When Single Cell Meets Long Read Sequencing
At a glance:
- What is Single-Cell Long-Read Sequencing?
- Analysis of single cell long read long RNA-seq data
- Application of single-cell long read long sequencing in tumor research
- Technological challenges and breakthroughs
- Future prospects and application prospects
- Conclusion
Recent years have witnessed remarkable progress in cellular-level genomic analysis techniques, revolutionizing our approach to cancer research. While traditional short-read approaches have provided valuable insights, their limitations in detecting transcript variants and analyzing gene expression patterns in neoplastic cells have restricted our understanding of cancer-related transcriptional dynamics. Emerging technologies for long-read sequencing, particularly through innovations from PacBio and Oxford Nanopore platforms, now facilitate comprehensive examination of complete transcripts at individual cell resolution. By surpassing the constraints of earlier methodologies, these advanced platforms enable detailed characterization of both transcriptional variants and genetic modifications. The enhanced capabilities for investigating cellular heterogeneity and microenvironmental factors within tumors mark this technology as transformative for oncological research. This methodological advancement provides researchers with unprecedented tools to explore cancer biology, offering deeper insights into the molecular mechanisms underlying malignant progression. Such developments represent a significant step forward in our capacity to decode the complexities of cancer development and progression.
What is Single-Cell Long-Read Sequencing?
Long-read sequencing at the single-cell level represents a sophisticated integration of two key methodologies: individual cell analysis and extended-read DNA sequencing approaches. This cutting-edge technique enables researchers to conduct detailed examinations of genomic, transcriptomic, and epigenetic characteristics within isolated cells. The technology's capability to sequence complete transcripts from individual cells provides enhanced precision in identifying alternative splice variants, uncovering genetic polymorphisms, and investigating modifications to the epigenome.
Limitations of short read sequencing
Traditional single-cell gene expression analysis methods rely mainly on short read and long sequencing technology. Although they can provide an overall landscape of the cell's transcriptome, they cannot reveal all the information in the full-length transcript, especially for long, highly variable transcript isoforms. This leads to the inability to accurately identify and quantify the expression levels of these isoforms, limiting a deeper understanding of cellular heterogeneity and complex transcription events.
Advantages of long read sequencing
Long-read sequencing technologies, such as Oxford Nanopore and PacBio, can provide complete transcript sequence information, solving the shortcomings of short read and long sequencing in the detection of transcript isoforms. By measuring the full-length sequence of each transcript, long-read-long sequencing can more accurately capture different transcriptional isoforms of genes, including details that may be missed in short-read sequencing. In addition, long-read sequencing can effectively reduce data noise caused by inaccurate transcript splicing, thereby improving data reliability and accuracy.
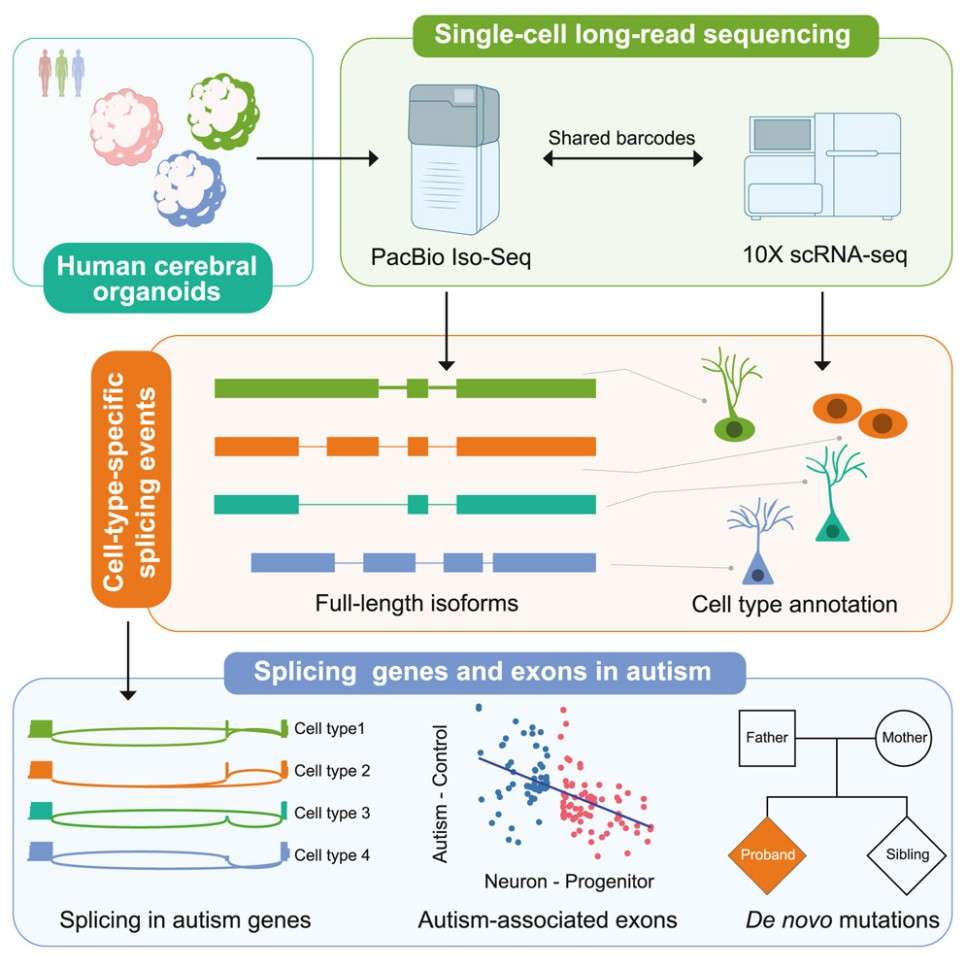 Figur1 . Single-Cell Long-Read Sequencing.(Yang, Y., et.al , 2023)
Figur1 . Single-Cell Long-Read Sequencing.(Yang, Y., et.al , 2023)
Analysis of single cell long read long RNA-seq data
Single-cell long-read long-RNA sequencing (scRiso-seq) technology provides in-depth understanding of gene expression and splicing isoforms by sequencing full-length transcripts from individual cells. Compared with traditional short-read long RNA sequencing, scRiso-seq can capture complete transcript information and reveal the isomeric diversity of transcripts. However, due to the high error rate of long-read and long-sequencing platforms, accurately assigning cell barcodes (CBs) and unique molecular barcode is a critical step in the analytical process.
Identification and correction of CBs
In scRiso-seq, accurate allocation of CBs and unique molecular barcode is critical for subsequent analysis. To this end, researchers have developed multiple bioinformatics tools that combine different library construction methods to correct and/or allocate CBs and unique molecular barcode. These tools can be divided into two categories:
Tools that rely on short reading of long data:
SiCeLoRe: Using short-read-long data as a whitelist for CBs and unique molecular barcode, combined with long-read-long data for comparison, the accuracy rate exceeds 97%.
Snuupy: improves SiCeLoRe, removes the reliance on polyA tails, and directly searches for CB and molecular barcode combinations in unaligned areas of long-read long data.
scNapBar: Based on SiCeLoRe, it reduces the reliance on the depth of short readings and long data and uses a naive Bayes model to predict the correctness of CB allocation.
Use only tools for long reading of long data:
Sockeye: is used to identify CBs and unique molecular barcode in Nanopore long-read data, but does not perform isoform level single-cell analysis.
BLAZE: uses a three-step approach to identify CBs, first determining the location of the barcode, then removing low-quality barcodes or not on the 10X whitelist, and finally using a quantile system to retain high-counted CBs.
In addition, scCOLOR-seq uses custom designed oligonucleotides to identify and correct CBs and molecular barcode through directional protocols and Levenshtein distances without using short read length data.
Single cell long read long RNA sequencing analysis process
After successfully allocating CBs and unique molecular barcode, subsequent analysis steps include:
Alignment and error correction: Long read data needs to be aligned to the reference genome, and error corrected for the splice sites read. This process can be done by referencing the genome or assembling transcripts. For example, long-read data generated using the Promethion sequencing platform can output approximately 80M of full-length sequences distributed to individual cells through Promethion's 3'gene expression library for multigene and isoform analysis. In addition, long-read and long-sequencing technologies (such as PacBio and Nanopore) can directly read full-length transcripts, reducing false positives, but require high integrity of long fragments. In some cases, short reads of long data may be used to assist in correcting errors in long reads of long data.
Transcript assembly: Long read data can be used to build complete transcripts and identify different splice isoforms. For example, the Nanopore platform combines 10X Genomics technology to generate high-quality single-cell full-length transcriptome sequences, supporting isoform detection and fusion gene analysis.
Quantitative analysis: Transcripts were quantitatively analyzed to assess their expression levels in different cells. For example, tools such as Seurat can be used to normalize gene or isoform counts and quantify their expression levels. In addition, potential biological changes can be identified by comparing expression levels under different conditions.
Differential transcript analysis: Compare differences in transcript expression under different conditions and identify potential biological changes. For example, through differential expression analyses (such as edgeR or DESeq2), transcripts that change significantly under different conditions can be identified. In addition, cell heterogeneity can be visualized using dimension reduction methods such as UMAP.
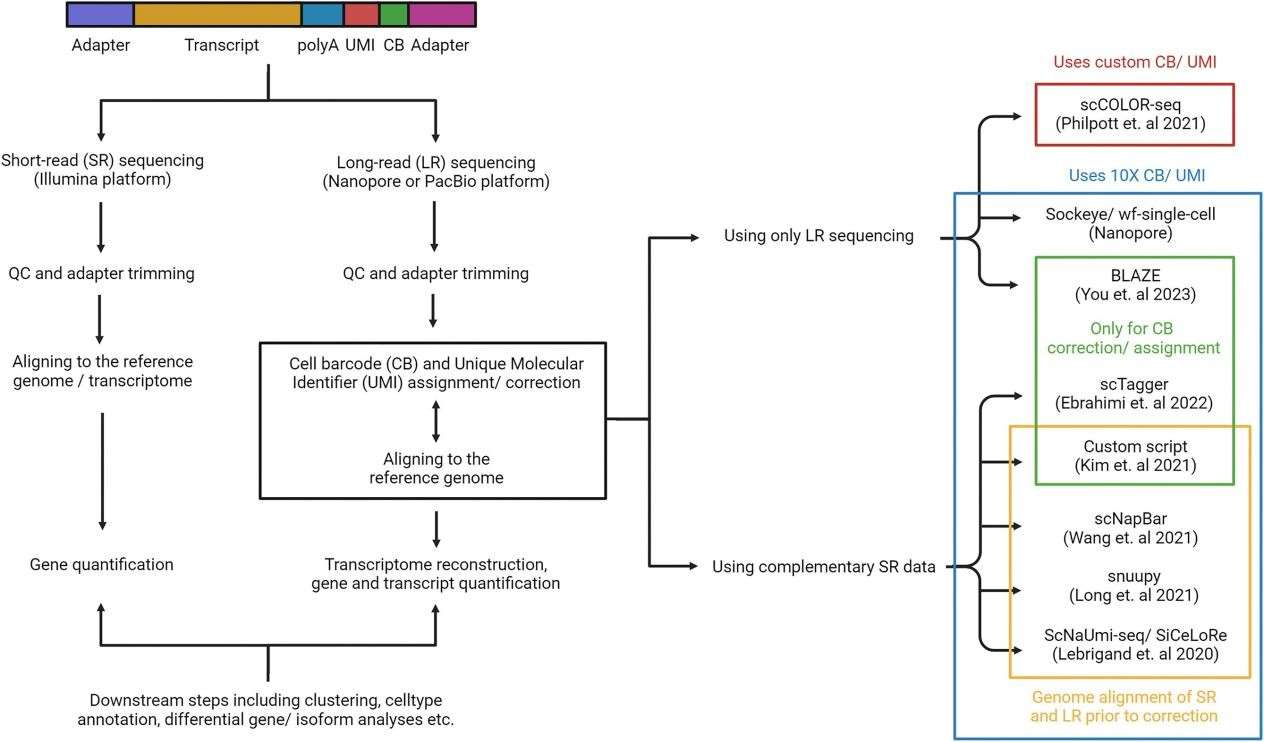 Figur2 . A typical long-read single-cell RNA sequencing workflow.(Gupta, P., et.al , 2024)
Figur2 . A typical long-read single-cell RNA sequencing workflow.(Gupta, P., et.al , 2024)
Application of single-cell long read long sequencing in tumor research
Single cell long read long sequencing in ovarian cancer research
In recent years, single-cell long read long sequencing technology has made breakthrough progress in the research of ovarian cancer and other tumors. Using the scTaILoR-seq technology, researchers were able to analyze transcriptional variations in ovarian cancer at single-cell resolution. This technology combines the advantages of single-cell RNA sequencing and long-read-long-sequencing. It not only accurately detects transcript isoforms in tumor cells, but also reveals differences in transcript expression between cell populations, thereby providing insight into tumor heterogeneity and its impact on cancer development.
Analysis of transcriptional variants in ovarian cancer by scTaILoR-seq technique
In ovarian cancer research, the scTaILoR-seq technology helped researchers reveal transcript isoforms in tumor cells and measure the relationship between expressed single nucleotide variants (SNVs) and transcript structure through single-cell resolution. This discovery not only helps us understand how tumor cells evolve at the molecular level, but also provides a theoretical basis for individualized treatment strategies.
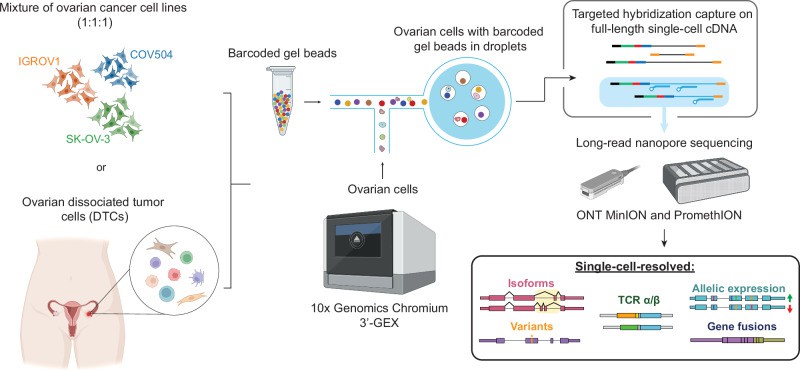 Figur3 . Single-Cell Long-Read Sequencing.(Byrne, A., et.al , 2024)
Figur3 . Single-Cell Long-Read Sequencing.(Byrne, A., et.al , 2024)
Revealing cellular heterogeneity in the tumor microenvironment
Cellular heterogeneity in the tumor microenvironment has a profound impact on the occurrence, development and treatment response of cancer. Long-read-long sequencing can finely depict the transcript changes of tumor cells in different microenvironments, providing a new perspective for studying the cellular heterogeneity of tumor microenvironments. By performing single cell analysis of different types of tumor cells, researchers were able to reveal transcription differences between cells and further explore their association with cancer immune escape, metastasis and drug resistance.
Transcript isoforms and gene regulation revealed by long-read and long-sequencing
Transcript isoforms play an important role in the occurrence and development of cancer. Long-read-long sequencing technology can reveal the expression patterns of different transcript isoforms in tumor cells, thereby providing important clues for studying the molecular mechanism of cancer. For example, certain transcript isoforms may be closely related to cancer cell proliferation, migration and resistance to treatment. Long-read-long sequencing can accurately capture these changes and provide data support for early diagnosis, prognosis assessment and precise treatment of cancer.
Technological challenges and breakthroughs
Data processing and technical challenges
Although long-read and long-sequencing technology has huge application potential, its application in tumor research still faces a series of challenges. First of all, the amount of data generated by long-read and long-sequencing is huge, which requires strong computing power and data storage capabilities. Secondly, due to the low sequencing depth, there may be more artifacts in long-read and long-sequencing, which may affect the accuracy and reliability of the data. In addition, complex transcript structures and highly heterogeneous cell populations make data analysis extremely difficult.
Technological breakthroughs and solutions
To solve these technical challenges, researchers have developed a variety of innovative methods. For example, scTaILoR-seq technology improves the sensitivity and efficiency of sequencing through a target enrichment strategy, thereby effectively solving the problem of low throughput for long reads and long sequencing. In addition, the continuous optimization of data analysis methods, such as splicing algorithms, noise reduction technologies and the application of deep learning models, makes the analysis of long-read and long-sequencing data more accurate and efficient.
Future prospects and application prospects
The extensive applications
With the continuous maturity of single-cell long read long sequencing technology, its application prospects in cancer research are very broad. In addition to ovarian cancer, single-cell long-read-long-sequencing technology is expected to play an important role in multiple tumor types, including lung cancer, breast cancer, gastric cancer, etc. In addition, this technology can also be combined with other high-throughput technologies (such as single cell genomics, spatial transcriptomics, etc.) to provide more accurate data support for early diagnosis, personalized treatment and efficacy monitoring of cancer.
From basic research to clinical application
With the advancement of technology, single-cell long read long sequencing is expected to move from basic research to clinical application. In early diagnosis of cancer, single-cell long read long sequencing can help discover early mutations and transcript mutations, providing an important basis for early screening of tumors. In addition, this technology can also be used to monitor efficacy, help doctors evaluate the effectiveness of treatment options, and adjust treatment strategies in a timely manner when patients develop drug resistance.
The direction of future technological development
In the future, single-cell long-read-long-sequencing technology may achieve further breakthroughs in sequencing throughput, accuracy and cost. With the continuous optimization of technology, we can foresee that long-read and long-sequencing will play an increasingly important role in cancer research, immunology research and other biomedical fields.
Conclusion
Single-cell long-read sequencing technology integrates single-cell sequencing with long-read sequencing, overcoming the limitations of short-read sequencing in detecting transcript isoforms. This capability allows for the comprehensive capture of full-length transcript sequences, offering significant advantages in studying complex transcriptional changes and gene expression variations, particularly in cancer research. By enabling precise analysis of transcript isoforms and genetic variations within tumor cells, single-cell long-read sequencing provides a valuable tool for exploring cell heterogeneity, the tumor microenvironment, and their roles in cancer development. Despite challenges such as data processing and sequencing depth, ongoing advancements in related technologies are expected to enhance the utility of single-cell long-read sequencing, positioning it as an increasingly crucial technique in fundamental research.
References
- Yang, Y., Yang, R., Kang, B., et.al. (2023). Single-cell long-read sequencing in human cerebral organoids uncovers cell-type-specific and autism-associated exons. Cell reports, 42(11), 113335. https://doi.org/10.1016/j.celrep.2023.113335
- Gupta, P., O'Neill, H., Wolvetang, E. J., Chatterjee, A., & Gupta, I. (2024). Advances in single-cell long-read sequencing technologies. NAR genomics and bioinformatics, 6(2), lqae047. https://doi.org/10.1093/nargab/lqae047
- Byrne, A., Le, D., Sereti, K., Menon, H., et.al. (2024). Single-cell long-read targeted sequencing reveals transcriptional variation in ovarian cancer. Nature communications, 15(1), 6916. https://doi.org/10.1038/s41467-024-51252-6
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment