


We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Overview of The Genome Assembly
- What is Genome Assembly
- De novo genome assembly
- Challenges and Solutions
- Technologies and Algorithms for DNA Sequencing
- Data Preprocessing
- Case Study of Genome Assembly
- Applications and Future Directions
- Conclusion
Genome assembly is an essential tool in contemporary genomics , allowing scientists to build entire genome sequences from the raw sequencing data. It is critical for understanding biological processes, evolutionary kinship, and apart from giving insights into the genetic components of the diseases. A near-complete genome assembly serves as a detailed map of an organism's genetic blueprint, enabling various applications from evolutionary biology to precision medicine. Genome assembly is a complex and challenging process, despite its transformative power, as the structure of genomes often involves repetitive sequences, long intergenic regions, and sequencing errors. These developments render genome assembly not merely a technical achievement, but a critical instrument in deciphering the secrets of life.
What is Genome Assembly
Genome assembly is the process of assembling millions or billions of short DNA fragments, known as reads, into contiguous sequences that represent the organism's genome. This is similar to piecing together an enormous jigsaw puzzle without an entire reference picture. The end-goal of genome assembly is an accurate, gapless reconstruction of the genome at chromosomal-level resolution if possible. This task is complicated by biological characteristics (genome size; repeat content; heterozygosity) and technical constraints imposed by sequencing platforms.
Service you may intersted in
De novo genome assembly
In genome assembly, at its core, it requires an understanding of its basic building blocks and approaches:
- Contig and Scaffold: A contig is, broadly speaking, a stretch of DNA reconstructed from overlapping sequencing reads. Contigs are the components of the genome assembly process. Contigs are joined together into scaffolds (with gaps representing unresolved regions) using additional data from mates in mate-pair sequencing, long reads or optical maps. Scaffolds lend a degree of higher-order structure, approximating the chromosomal architecture.
- De novo vs. Reference-guided Assembly: De novo genome assembly constructs genomes entirely from raw sequencing reads without relying on prior genomic information. This method is particularly valuable for studying organisms with no available reference genome or for exploring the full spectrum of genetic diversity in non-model species. De novo assembly uses computational algorithms to piece together overlapping reads into contiguous sequences. While it provides unbiased insights into a genome's structure, it is computationally intensive and requires high-quality, high-coverage sequencing data to minimize gaps and errors. Technologies such as PacBio and Oxford Nanopore, which generate long reads, are especially advantageous for de novo assembly due to their ability to span repetitive regions and resolve complex genomic structures. Reference-guided genome assembly, on the other hand, aligns sequencing reads to an existing reference genome, using it as a scaffold to assemble the target genome. This method is significantly less computationally demanding and faster than de novo assembly, making it suitable for organisms closely related to a well-characterized reference species. Reference-guided assembly excels in accurately reconstructing known genomic regions and identifying small-scale variants, such as single nucleotide polymorphisms (SNPs). However, its reliance on the reference genome introduces biases, potentially missing novel sequences, large structural variations, or unique genomic features of the target organism. Both methods have specific applications based on the research goals and the organism under study. For example, de novo assembly is critical in biodiversity studies where novel species are analyzed, while reference-guided assembly is often employed in clinical research to study human genomes and their variants. Hybrid approaches, which combine elements of both methods, are also emerging as powerful tools. By integrating de novo and reference-guided strategies, researchers can achieve high-resolution assemblies that capture both conserved and novel genomic features.
Challenges and Solutions
DNA sequence assembly had its own challenges; new techniques were had to solve these problems:
- Repetitive Sequences: Repeats make assembly difficult due to multiple possible mappings of reads. Such repeats, which are prevalent in eukaryotic genomes, can result in fragmented assemblies. Long-read sequencing platforms (such as those commercially available from PacBio and Oxford Nanopore) enable spanning these regions and even resolving more complex repeats. Such assembly accuracy is complemented by computational tools that annotate and mask repetitive sequences.
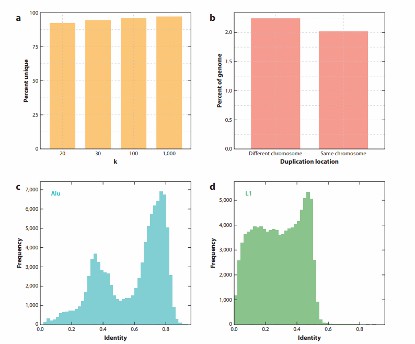 Repetitive content creates a challenge in genome assembly, as illustrated by the repetitive content of the human genome (Rice ES et al., 2018).
Repetitive content creates a challenge in genome assembly, as illustrated by the repetitive content of the human genome (Rice ES et al., 2018).
- Error and Complexity Management: Long reads are often impaired by sequencing errors that propagate through the assembly process. Highly accurate assemblies rely on post-assembly correction tools (e.g. Pilon for short-read polishing, Racon for long-read correction). Further, the raw computational resources necessary to assemble a genome are non-trivial and loaded with infrastructure-level solutions — spanning both an increasing reliance on the cloud to deliver these capabilities, as well as parallelized algorithms that can tackle these challenges head-on.
- Increased Vertebrate Genome Publications: As a result of these new technologies, the number of published vertebrate genomes has increased greatly in the past decade. This surge reflects the enhanced ability to sequence and assemble complex genomes, contributing to significant advances in fields such as comparative genomics, evolutionary biology, and biodiversity conservation.
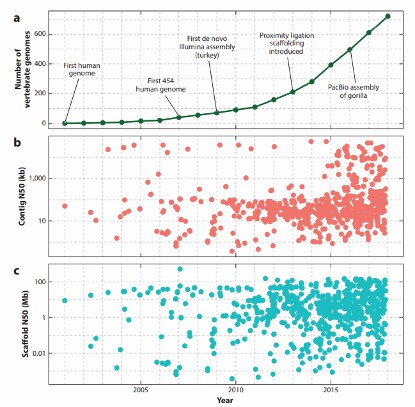 Timeline and statistics of vertebrate genome assemblies deposited in the National Center for Biotechnology Information's Genbank (Rice ES et al., 2018).
Timeline and statistics of vertebrate genome assemblies deposited in the National Center for Biotechnology Information's Genbank (Rice ES et al., 2018).
Technologies and Algorithms for DNA Sequencing
Genome assembly tools
The sequencing technologies and computational algorithms play a critical role in any genome assembly project. These tools have developed rapidly into the unprecedented ability to tackle even complex genomes.
Sequencing technologies can be classified according to read length, accuracy, and throughput:
Generally, the second-generation Sequencing (SGS) platforms lead with this category (short reads 50–300 bp) which yield high throughput, cost-effectiveness, and superior quality, Illumina. Short reads make depth of coverage by design, which is essential for correcting errors and resolving small-scale genomic features. Indeed, their short length limits their coverage of repetitive or complex-structured regions.
Third-generation Sequencing (TGS) include technologies like PacBio and Oxford Nanopore that produce long reads, often tens of kilobases or longer. Such reads are essential for resolving repetitive regions, characterizing structural variations, and producing higher-contiguity genome assemblies. Within its error rates are broadly higher than those of SGS, TGS chemistry and computational error correction have been highly developed to enhance data quality.
The combination of SGS and TGS captures the benefits of both short and long reads: short reads guarantee baseline accuracy at the base level, while long reads increase contiguity and structural resolution. Overcoming the limitations of each technology, hybrid approaches have become the norm for constructing complex genomes.
The process of converting input genomic DNA into sequencing libraries is necessarily platform dependent.
 Overview of sequencing library architecture, output, and assembly results from three high-throughput sequencing technologies (Rice ES et al., 2018).
Overview of sequencing library architecture, output, and assembly results from three high-throughput sequencing technologies (Rice ES et al., 2018).
Genome assembly in bioinformatics
Algorithms reconstruct sequences using graph-based structures and statistical models in genome assembly:
- Graph-based Methods: De Bruijn Graph (DBG) based approaches fracture the reads into k-mers, creating a graph with k-mers as nodes and sequence reconstruction paths as potential genomic sequences. DBG is computationally efficient and well-optimized for short-read data but performed poorly against high-error environments and repeats. Overlap-Layout-Consensus (OLC) methods, however, are intended for long reads, alignments of full sequences to find overlaps and create layouts. OLC is good for complex genomes, but it is more computationally intensive.
- Integrated Tools: Integrated approaches Modern assemblers such as SPAdes and MaSuRCA combine aspects of the DBG and OLC frameworks that provide optimal performance on hybrid datasets. These tools take advantage of the benefits from both short- and long-read technologies, resolving the weaknesses found in either technology to create high-quality assemblies.
- Steps in Genome Assembly: Genome assembly requires passing through a number of well-established stages that must be successfully carried out for optimal results. These involves preprocessing, assembly and classifying quality assessment.
Data Preprocessing
The preprocessing step makes sure that our input data is clean and ready to be assembled in the first place:To help you maintain your work and ensure high-quality sequence analysis output, for example, fastQC will help you evaluate the quality of sequencing reads, low-quality regions, adapter contamination, and other artifacts Note that cleaning these data improves downstream assembly performance.
There is information on your second oneTrimming and Filtering: These tools, including Trimmomatic and Cutadapt for instance, remove adapter sequences as well as low-quality bases from the reads. Filtering limits the impact of contaminants and sequencing error, with only high-confidence reads represented in the assembly.
Read normalization adjusts coverage across the genome, which can help reduce biases introduced by over-represented regions. This step is critical for limiting computational burdens in high-coverage datasets.
Genome assembly steps
The core assembly process consists of an iterative cycle:
This process of contig construction, more or less, involves arranging all the reads into contigs that are the best-guess sequences of the same genomic region without requiring external data. Specialised tools exist for this phase (e.g. Canu for long reads or Velvet for short reads).
- Scaffolding and Gap Filling: Scaffolding assembles contigs into more extensive structures, using long-read or mate-pair data, and gap-filling software, like GapCloser, attempts to fill in missing sequences. These steps also lead to higher-level completions of assemblies and higher accuracy in assemblies.
- Error Correction: Before the assembly is complete, it is passed through site-specific error correction tools such as Pilon or Racon, which corrects base errors and debts misassemblies through these tools. To ensure good results for downstream tasks this step is vital.
Quality Assessment
Quality assessment confirms the assembly is trustworthy and complete:
Metrics: N50 is a common metric describing contiguity of assembly; BUSCO assesses completeness by checking presence of conserved genes.
Validation Metrics: QUAST produces detailed reports of assembly statistics, highlighting errors, misassemblies, and areas needing additional improvement. Instead, REAPR aims to identify structural inconsistencies and highlight where assemblers can improve.
Genome Annotation
Genome assembly is only the first step toward understanding the biological functions encoded within a genome. Genome annotation involves identifying genes, regulatory elements, and functional regions within the assembled sequences. This step transforms raw sequences into a biologically meaningful framework:
- Structural Annotation: Involves identifying genomic features such as protein-coding genes, non-coding RNAs, promoters, and introns. Tools like AUGUSTUS and MAKER automate the prediction of gene models by integrating sequence data with transcriptome evidence and known protein sequences.
- Functional Annotation: Assigns biological roles to genomic features by linking them to existing databases such as GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), and UniProt. Tools like InterProScan and BLAST are commonly used to align predicted proteins to annotated sequences, providing insights into their potential functions.
- Challenges in Annotation: Annotation accuracy depends on the quality of the assembly and the availability of reference data. Poorly assembled regions, such as those containing repeats or gaps, can lead to incomplete or incorrect annotations. For non-model organisms, the lack of well-curated reference datasets poses additional challenges.
- Automation and Manual Curation: Automated pipelines streamline the annotation process but often require manual curation to verify predictions and resolve discrepancies. Collaborative platforms like
Case Study of Genome Assembly
Background
The wheat genome is one of the most complex plant genomes due to its large size, hexaploid nature (three homologous sets of chromosomes), and high repeat content. Wheat is a staple crop worldwide, making its genetic understanding critical for improving agricultural yields, disease resistance, and climate resilience. Decoding its genome posed a significant challenge to researchers, requiring a combination of advanced sequencing technologies and computational approaches.
Methods
To tackle this complexity, researchers employed:
- Sequencing Technologies: A hybrid approach was used, combining second-generation sequencing (short reads from Illumina) with third-generation sequencing (long reads from PacBio and Oxford Nanopore). Optical mapping and chromosome flow sorting were also employed to anchor scaffolds and resolve chromosome structures.
- Assembly Algorithms: Tools like Canu and Hi-C scaffolding software were utilized to generate highly contiguous assemblies. The De Bruijn graph method facilitated the assembly of repetitive regions.
- Validation: The assembly was validated using BUSCO to measure gene completeness and alignment-based methods to confirm accuracy.
Results
The assembly achieved a high-quality reference genome for wheat, covering over 90% of the genome with unprecedented resolution. Key genes associated with yield improvement, disease resistance (e.g., rust resistance), and environmental stress tolerance were identified. This genome assembly enabled precision breeding strategies, improving wheat resilience to global climate challenges.
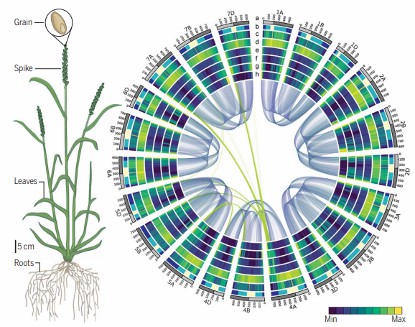 Wheat genome deciphered, assembled, and ordered (International Wheat Genome Sequencing Consortium (IWGSC) 2018).
Wheat genome deciphered, assembled, and ordered (International Wheat Genome Sequencing Consortium (IWGSC) 2018).
Applications and Future Directions
Applications
Genome assembly has applications across various fields:
- Model Organisms: High-quality assemblies for model organisms like mice and zebrafish underpin genetic, developmental and comparative investigations. These assemblies act as reference standards to allow accurate annotation and functional exploration of genes.
- Non-model Organisms: De novo genome assemblies have provided insight into biodiversity, adaptation and ecological interactions. As an example, the assembly of genomes of economically important crops like wheat and rice has enabled the identification of traits to improve yield and stress tolerance. Data on a genome of an endangered species, for example, enables greater insights into genetic diversity and informs breeding programmes for conservation.
- Clinical Research: Genome assemblies have important applications in precision medicine, allowing for the identification of genetic factors contributing to diseases and the potential for the development of targeted therapies. Genome assemblies are used in cancer genomics for the identification of structural variations and mutations driving tumorigenesis. For example, pathogen assemblies like SARS-CoV-2 have fast-tracked both vaccine development and epidemiological tracking.
Future Directions
Trends and emerging technologies hold promise for even more sophisticated genome assembly:
- Ultra-long reads: Technologies generating reads longer than 1 Mb allow for the assembly of previously intractable regions of the genome, including centromeres and telomeres. Promise chromosome-level assemblies for complex genomes.
- AI-Powered Assembly: The machine learning algorithms aiding in error correction, repeat resolution and structural variation detection.
- Tracking Genetic Heterogeneity: Single-cell methods gain important insights around genetic heterogeneity but allow for subsequent haplotype-specific reconstruction, affecting population genetic studies and furthering paleo-/ancient DNA studies and personalized medicine.
- Data Standardization and Sharing: Open data repositories and standardized pipelines that allow contributors to upload their data can help facilitate reproducibility and collaboration across the scientific community.
Conclusion
Genome assembly is a fundamental resource in contemporary biology that provides unparalleled information on the structure, function and evolution of genomes. Rapidly improved sequencing technologies, algorithms and computational systems have made genome assembly a more efficient and more accessible process. Next-generation genome assembly techniques are expected to broaden in their scope and impact due to innovations such as ultra-long-read sequencing, AI-based methodologies, and single-cell assembly methodologies. These advancements will further influence disciplines ranging from medicine to agriculture to conservation, propelling transformative progress in our ability to understand and utilize the blueprint of life.
References
- Rice, E. S., & Green, R. E. (2019). New Approaches for Genome Assembly and Scaffolding. Annual review of animal biosciences, 7, 17–40. https://doi.org/10.1146/annurev-animal-020518-115344
- International Wheat Genome Sequencing Consortium (IWGSC) (2018). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science (New York, N.Y.), 361(6403), eaar7191. https://doi.org/10.1126/science.aar7191
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment