

We are dedicated to providing outstanding customer service and being reachable at all times.
The Basics and Beyond of PacBio SMRT Sequencing
At a glance:
- What is PacBio sequencing?
- How does PacBio sequencing work?
- What is HiFi Read? How does PacBio HiFi work?
- What are the differences between Illumina and PacBio?
- What is the difference between PacBio and Nanopore?
- What are the benefits of PacBio sequencing?
- What are the applications of PacBio sequencing?
What is PacBio sequencing?
PacBio sequencing platform is a single-molecule real-time sequencing technology developed by Pacific Biosciences. PacBIo sequencing features long read length, high accuracy, uniform genome coverage, and detection of base modifications while sequencing.
How does PacBio sequencing work?
PacBio sequencing technology is based on the principle of sequencing while synthesizing, with sequencing lengths up to 30kb and throughputs up to 20 Gb. PacBio sequencing captures sequence information during the replication process of the target DNA molecule. The template, called a SMRTbell, is a closed, single-stranded circular DNA that is created by ligating hairpin adaptors to both ends of a target double-stranded DNA (dsDNA) molecule.
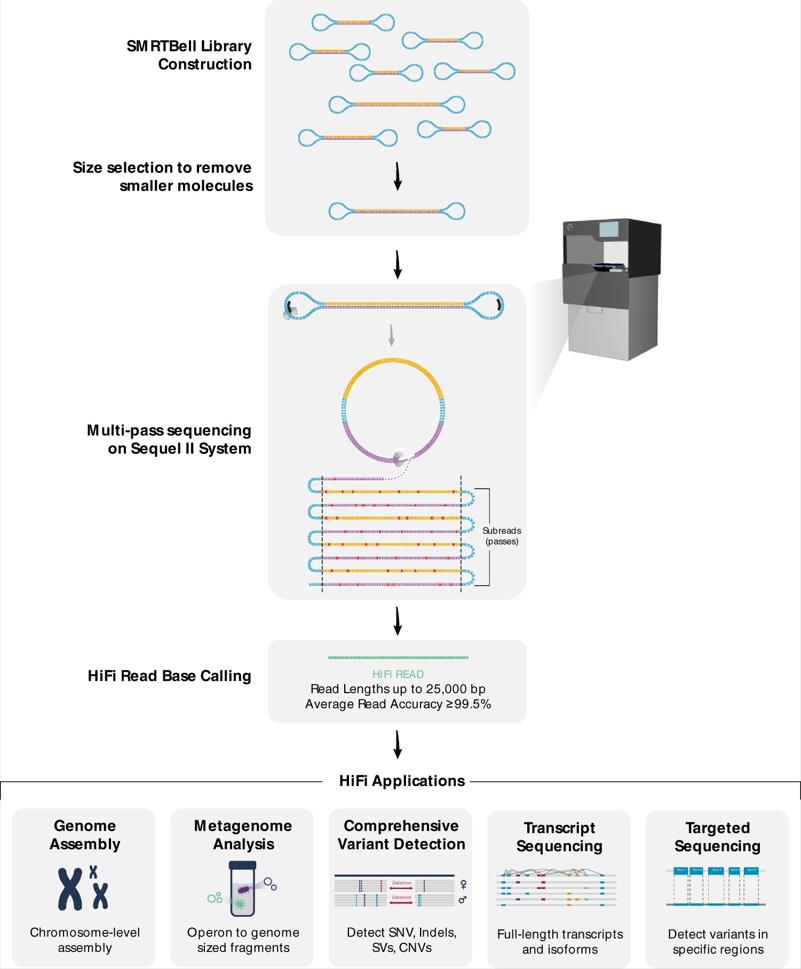 Workflow of PacBio Sequencing (Hon T et al. 2020)
Workflow of PacBio Sequencing (Hon T et al. 2020)
What is SMRT sequencing?
Single Molecule, Real-Time sequencing, which we abbreviate as SMRT sequencing, is a technology introduced by Pacific Biosciences of California, Inc. (PacBio).
SMRT sequencing uses four-color fluorescently labeled dNTP and ZMW wells to complete the sequencing of a single DNA molecule. In each ZMW well, a single DNA molecule template is bound to a primer and then, after binding DNA polymerase, is immobilized to the bottom of the ZMW well. When four-color fluorescently labeled dNTP is added and DNA synthesis begins, the attached dNTP will stay at the bottom of the ZMW for a longer period of time due to base pairing and emit a corresponding fluorescent signal after excitation to be recognized, and the returned fluorescent signal will form a special pulse wave. On the other hand, because the fluorescence signal is attached to the phosphate group of dNTP, when the last dNTP is synthesized, the phosphate group is automatically shed, which ensures the continuity of the detection and improves the detection speed of 3 bases per second synthesized with a high-resolution optical detection system, real-time detection is achieved.
What is HiFi Read? How does PacBio HiFi work?
HiFi reads, or High Fidelity Long Reads for short, is based on Circular Consensus Sequencing (CCS) mode to produce both long read length (10-20kb length) and high accuracy (>99% accuracy) sequencing results.
PacBio HiFi sequencing is currently the model for excellent data types for a variety of genomic applications. In this sequencing mode, the enzyme read length is typically larger than the insert length, so the enzyme is sequenced in a rolling loop around the template and the insert is sequenced multiple times. Random sequencing errors caused during a single sequencing can be corrected by the algorithm itself, resulting in highly accurate HiFi reads.
What are the differences between Illumina and PacBio?
- The sequencing principles are different. The next-generation sequencing is a high-throughput sequencing technology developed based on PCR, while long-read sequencing uses a reversible termination end, which can realize sequencing while synthesizing.
- The difference of library building process. Taking genomic DNA as an example (6-20k libraries), the library-building process is similar to "short sequence massively parallel sequencing", both of them fragment the genome and then add specific splice sequences at both ends of the fragmented DNA, the main difference is that the final bell library needs to be combined with sequencing universal primers, strand-swapping The main difference is that the final bell library needs to be sequenced with universal primers, DNA polymerase with strand substitution, forming a complex of template, primer and polymerase to be tested, and then loaded onto the sequencing chip.
- Unlike sequencing libraries, SMRTbell libraries are single-stranded hairpin connectors, and the connectors attached to both ends of the library are identical (except for barcode sequences in Dual index). This is due to the principle of rolling-loop sequencing, which does not require a partially complementary, partially free structure like Illumina or MGI adapters to achieve PCR and double-end sequencing.
- PacBio provides two types of splice sequences, one is the familiar A/T sticky end splice, and the other is the flat end splice, which is used when the insert fragment is larger than 250bp, and vice versa (which of course means that in 99% of cases, the flat end is used).
- Different fluorescent moieties. Unlike existing second-generation sequencing technologies that label the fluorescent moiety on the 5' end methyl group, the fluorescent moiety of dNTP in SMRT sequencing is labeled on the phosphate group, which is subsequently shed after the synthesis is completed, the same as the natural DNA strand synthesis product, ensuring the extra-long read length of sequencing.
- Illumina focuses on short-read sequencing, producing read lengths of 50-300 base pairs (bp), while PacBio HiFi sequencing can produce read lengths in excess of 10,000 base pairs. The advantage of long read lengths over the very time and labor-intensive assembly of short read data is that it is easier to perform and go all the way through the assembly and has advantages in identifying rare variants and large structural variants.
What is the difference between PacBio and Nanopore?
Both are long-read sequencing techniques, Nanopore and PacBio also have many differences and discrepancies. Nanopore reads are much longer than PacBio, they can reach 330kbp in length, even exceeding 2Mb according to one report. Yield/cell is 245 Gb. It can be used for both DNA and RNA (without reverse transcription), and it can read methylated bases (and other modifications) directly (read).
| PacBio SMRT | Nanopore | |
| Principle of sequencing | Sequencing by synthesis/DNA polymerase | Electronic signals sequencing/exonuclease |
| Read length | 10-15 kb, up to 20 kb | 10-100 kb, up to 4 Mb |
| Read accuracy (%) | 88–90/99.9 (CCS) | 96–99 |
| Based per sample | 20-30 Gb | 6 Gb |
| Runtime (h) | 10–30 | 72 |
| Advantages | Long average read length; No amplification of sequencing Fragments; More accurate in isoform discovery |
Ultra-long read; Electronic sequencing; Portable; No amplification of sequencing fragments; Powerful in expression level quantification |
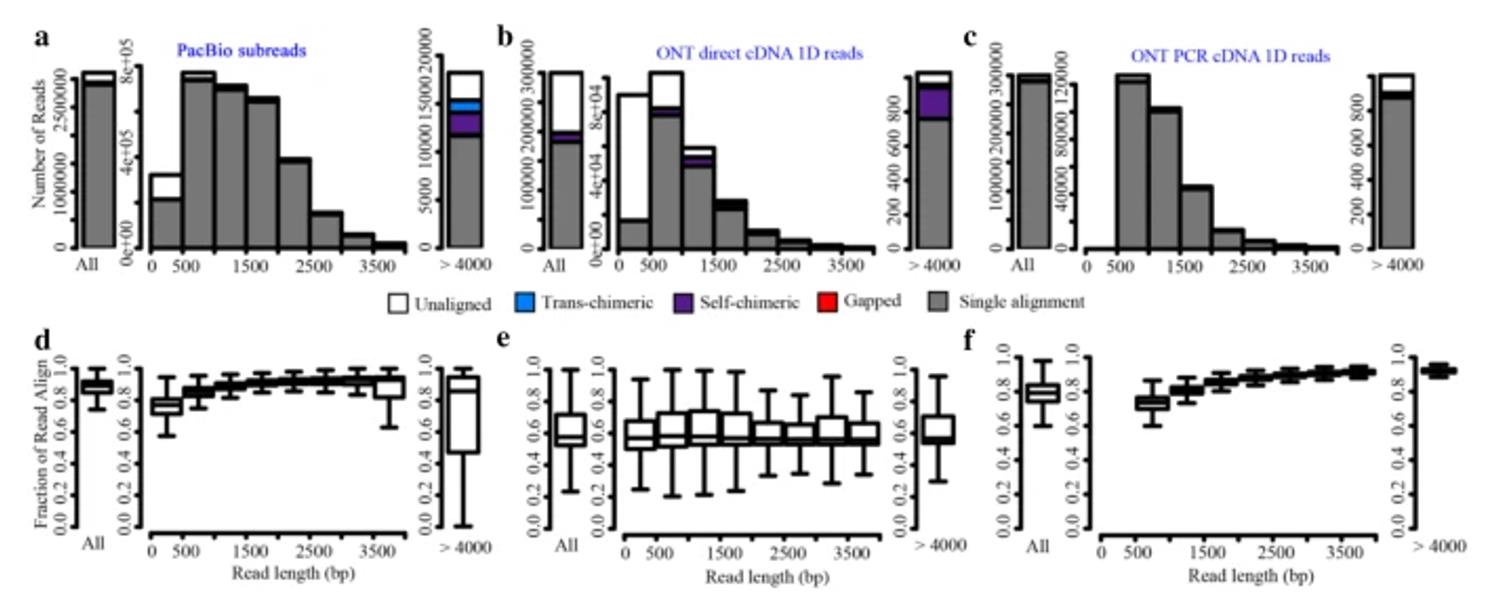 Length distributions and mappability of reads (Cui J et al. 2020)
Length distributions and mappability of reads (Cui J et al. 2020)
What are the benefits of PacBio sequencing?
PacBio sequencing confers four major advantages compared to other sequencing technologies: long read lengths, high consensus accuracy, a low degree of bias, and simultaneous capability of epigenetic characterization.
HiFi Reads means there is no need to choose between read length and accuracy. They are already widely used for 16S full-length amplicon sequencing, and genome assembly for metagenome contigs.
- Independent of GC preference and can accurately reveal diversity
Better assembly of macro-genomic data is possible, while DNA modification information can be directly detected. During base synthesis, tetra chromatically labeled dNTP releases a specific fluorescent signal and corresponds to a specific pulse signal. When a base carries a specific modification, the pulse signals of two adjacent bases appear with a corresponding time interval (Interpulse Duration, IPD), and the type of base modification can be determined based on the IPD value.
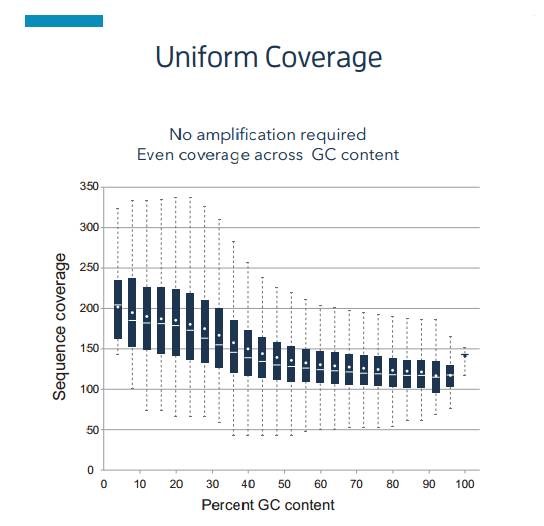
- Ultra-long sequencing read lengths
Using Sequel system to sequence human genome, 30kb fragments are built and run for 10h. The average read length is about 10~18kb, more than half of the reads are longer than 20kb, and the longest can reach 60kb, which is sufficient for most of the gene structure sequencing and achieve high quality assembly.
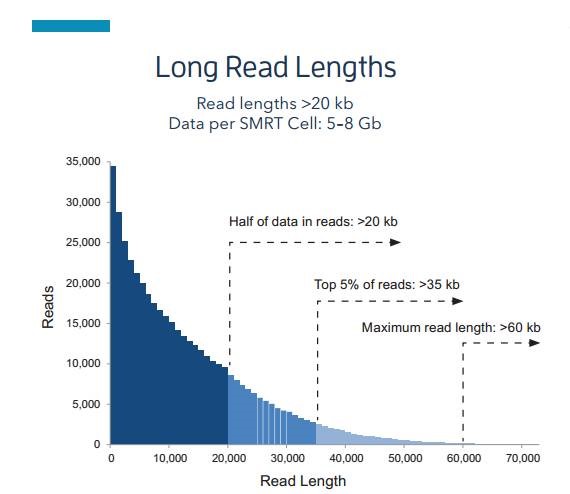
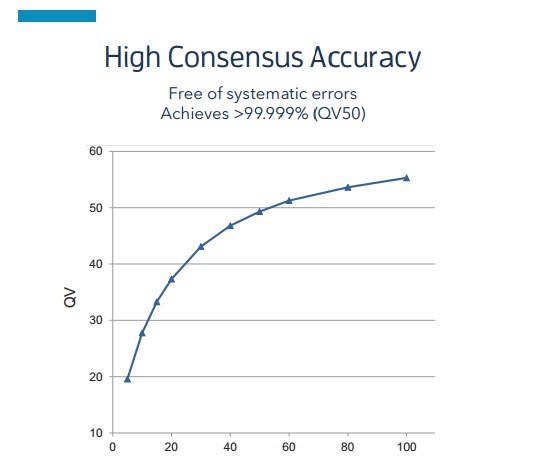
- Ultra-high accuracy
The chance of error in PacBio sequencing is random and independent of sequence length and sequence composition. This randomness tends to result in 87.5% accuracy at 1X for single molecule sequencing, but as shown in the figure, the accuracy of sequencing increases as the amount of sequencing increases. When the sequencing depth reaches 30X, the accuracy reaches Q50 (99.999%), and very accurate sequencing results can be achieved with 80X sequencing volume, while each reaction of the first and next generation sequencing is originally the average signal obtained from the simultaneous superimposed reactions of N molecules.
What are the applications of PacBio sequencing?
Whole genome sequencing and assembly
The complementary advantages of PacBio HiFi and ONT Ultra-long have improved the continuity and accuracy of genome assembly, laying a solid foundation for subsequent genome evolution, genome structure variation resolution and gene function research.
In particular, more and more species have completed T2T genome assembly, heralding the coming era of T2T genome explosion. The newly completed regions include all of the centromeric and telomere sequences, and for the first time, these complex regions of the genome are being used for variation and function studies.
Mutation detection
Reliable data covering a large range of genomes obtained by PacBio HiFi sequencing facilitates deep mining of genomic variants, such as SVs (genomic variants of 50 to 1,000 bp), and their classification and resolution. It also facilitates the study of algorithms for genotyping from short read-length data and corrects the errors in the existing perception of SVs.
Full-length transcriptome
Based on single-molecule real-time SMRT sequencing technology, Iso-Seq spans the complete transcript from the 5' end to the 3'-Poly A tail with the advantage of ultra-long read length to obtain high-quality full-length transcript sequences without interrupting RNA molecules. It was found that Iso-Seq has significant advantages in identifying novel transcripts, alternative splicing events and fusion gene studies.
Epigenetics
Another outstanding advantage of triple sequencing is the ability to directly read out base modifications (e.g. methylation), and the PacBio sequencing platform has now enabled the direct detection of 5mC methylation at CpG sites in DNA samples by HiFi sequencing. Therefore, we can obtain both accurate sequencing results and methylation information in the genome, and construct a genome-wide methylation map without additional experimental processing.
Microbiome strain identification
Most current techniques for microbiome strain typing, such as 16S rRNA sequencing or short-read sequencing, often provide insufficient resolution. A microbial species may only be classified as part of a broader genetic family, rather than being identified as a separate genetic species. PacBio SMRT sequencing technology, which captures ultra-long read lengths while also directly detecting base modifications, can help scientists to identify microbial strains. These base modifications can help the scientific community to address microbiome analysis methods for individual species and strains at high resolution.
References
- Cui J, Lu Z, Xu G, et al. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome[J]. Plant Methods, 2020, 16(1): 1-13.
- Hon T, Mars K, Young G, et al. Highly accurate long-read HiFi sequencing data for five complex genomes[J]. Scientific data, 2020, 7(1): 399.
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment