We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Challenges in Defining mRNA Transcript Boundaries Using Long-Read Sequencing
At a glance:
- Transcript Complexity and Isoform Diversity
- Sequencing Errors and Accuracy
- Coverage Variability and Expression Levels
- Contextual Influences and Structural Variants
- Algorithmic Approaches and Database Bias
- Comparing LRS Platforms: ONT vs. PacBio
The quest to decipher the intricacies of gene expression has taken a remarkable leap forward with the emergence of long-read sequencing technologies. By providing unprecedented insights into the full length of mRNA transcripts, these technologies promise to revolutionize our understanding of genetic regulation, alternative splicing, and isoform diversity. However, the road to unraveling the true beginning and end of mRNA transcripts is fraught with challenges that stem from the very nature of long-read sequencing data. In this article, we explore the multifaceted hurdles researchers face when identifying mRNA transcript start and end sites from long-read sequencing data.
Transcript Complexity and Isoform Diversity
At the heart of the challenge lies the sheer complexity of mRNA transcripts themselves. Genes can give rise to multiple isoforms through alternative splicing and differential promoter usage. Long-read sequencing enables the capture of full-length isoforms, which can be both a blessing and a challenge. The intricate landscape of isoforms can confound attempts to precisely determine where a transcript begins and ends. Multiple transcription start sites (TSS) and transcription termination sites (TTS) can coexist for a single gene, complicating the task of defining accurate transcript boundaries.
Sequencing Errors and Accuracy
While long-read sequencing platforms excel in generating longer reads, they are not immune to errors. Sequencing errors, which include insertions, deletions, and substitutions, can introduce inaccuracies during read alignment and affect the identification of start and end sites. These errors can cause misalignments, leading to incorrect annotations and potentially obscuring genuine transcript boundaries. Accurate identification demands sophisticated computational approaches capable of distinguishing genuine boundaries from sequencing artifacts.
Coverage Variability and Expression Levels
Unlike short-read sequencing, long-read sequencing data can exhibit variable coverage along the length of a transcript. This variability arises from differences in sequencing depth and transcript expression levels. Regions with low coverage might result in incomplete representation of transcript boundaries, introducing uncertainty into the annotation process. The uneven distribution of reads along a transcript poses challenges in distinguishing between genuine transcription boundaries and random variations in read coverage.
Contextual Influences and Structural Variants
Transcript start and end sites are not isolated events but are influenced by the three-dimensional chromatin conformation and interactions with regulatory elements. Long-read sequencing captures these nuances, but it also means that transcript boundaries might not be as clear-cut as previously assumed. Structural variations, such as gene fusions, deletions, and inversions, can further disrupt transcript structures, making boundary identification a complex puzzle. Accurate annotation necessitates the integration of long-read transcriptomics data with other genomic and epigenomic information.
Algorithmic Approaches and Database Bias
The development of robust algorithms for identifying transcript boundaries from long-read data remains an active research area. Algorithms designed for short-read data may not be directly applicable due to the unique challenges posed by long-read sequencing. The choice of alignment algorithms, error correction methods, and parameter settings significantly influences the accuracy of boundary identification. Furthermore, biases inherent in transcript annotation databases, often constructed using short-read data, can lead to misannotations or incomplete representations of transcript start and end sites in long-read data.
Comparing LRS Platforms: ONT vs. PacBio
The quest to identify RNA transcript start and end sites through LRS data is a journey rife with complexities. Variability across platforms, sample preparation methods, and technological limitations necessitate a holistic understanding of these challenges. As researchers delve deeper into the intricacies of transcriptomics using LRS, a cautious and multidimensional approach, encompassing rigorous experimental design, advanced analytical techniques, and meticulous quality control, is imperative.
Direct RNA sequencing entails the nanopore-based sequencing of natural RNA extracted from total or enriched samples after a selective synthesis of the complementary strand. This approach, while valuable, is prone to the effects of RNA instability, which can impact the accuracy of the generated data.
Another approach involves nanopore sequencing of full-length cDNA derived from PolyA+ RNA using a strand-switching protocol. This method, too, faces limitations associated with ONT sequencing technology accuracy, influencing the precision of results.
Iso-Seq sequencing, performed on the PacBio platform, relies on a strand-switching protocol to create full-length cDNA from PolyA+ RNA. The resulting cDNA is transformed into a dumbbell-shaped SMRTbell library structure for sequencing reactions. Through cyclic consensus sequencing, methodological error correction is implemented during the sequencing process.
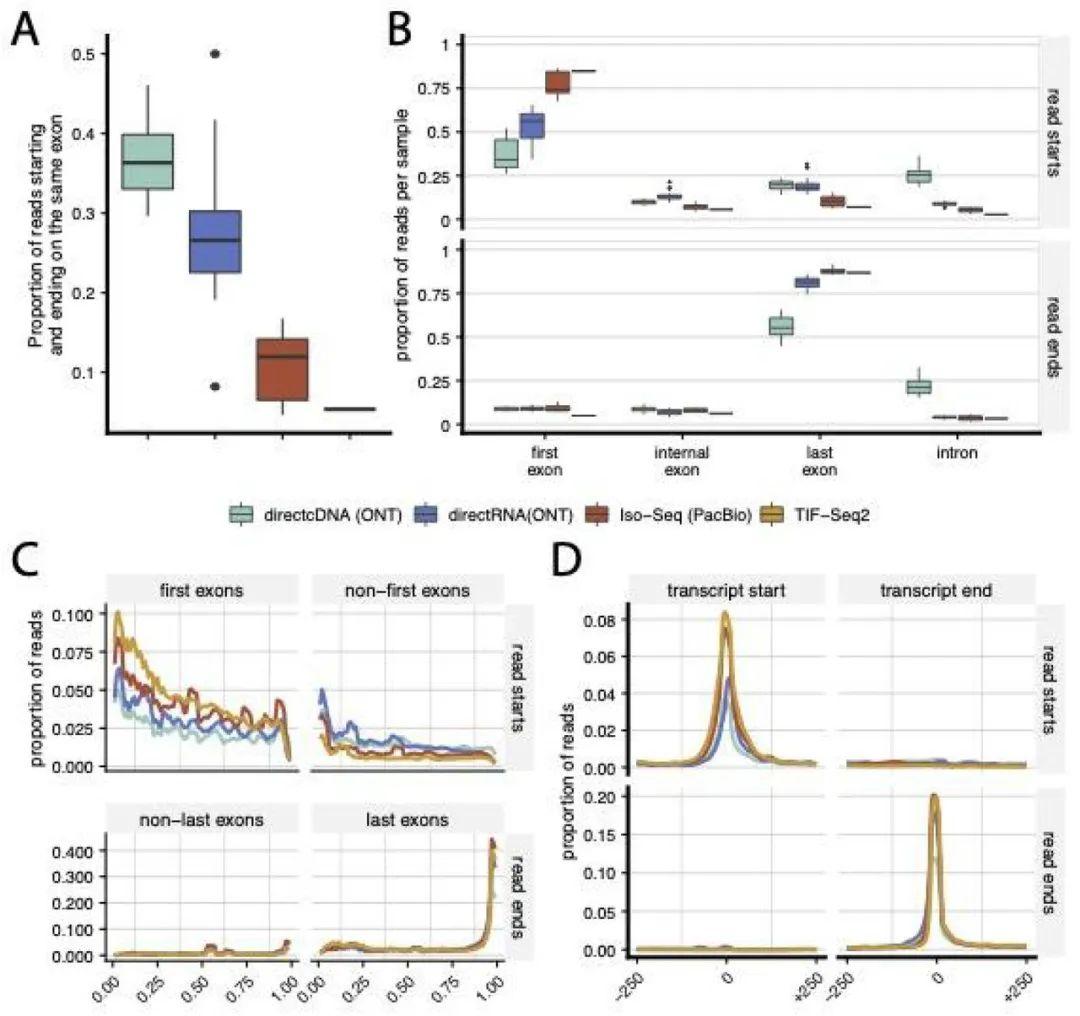 Relationship of the ends of LRS reads to the annotated ends of mRNA. (Calvo-Roitberg et al., 2023)
Relationship of the ends of LRS reads to the annotated ends of mRNA. (Calvo-Roitberg et al., 2023)
- Discrepancies and Challenges
Upon analysis, the study exposed a consistent observation: the start and end positions of transcript reads across different LRS methods often deviated from the 5' or 3' ends of annotated or empirically derived mRNA molecules. This discrepancy was particularly pronounced in direct RNA sequencing data from the ONT platform, resulting in frequent 5' truncations. Such truncations can have profound implications, potentially leading to incorrect identification of transcription start sites or first exons.
- Navigating for Solutions
The strength of LRS lies in its potential to unveil new isoforms, yet a cautious approach is necessary to mitigate the uncertainties arising from discrepancies in boundary identification. Filtering strategies may be employed to retain only those transcript reads that align with known 5' or 3' mRNA ends, enhancing confidence in biological findings. However, it is important to acknowledge that such filtering can come at the cost of significantly reducing the number of retained reads.
- Factors Influencing Variation
Numerous factors contribute to the observed discrepancies in transcript start and end site identification across LRS platforms and methods. The quality of RNA prior to library construction, reverse transcription inefficiencies, and challenges inherent to sequencing technology itself—such as nanopore-induced truncations—all play pivotal roles. Elements like single-strand speed, current fluctuations, and motor protein behavior in nanopore sequencing can all contribute to read truncations and inaccuracies.
Reference
- Calvo-Roitberg, Ezequiel, Rachel F. Daniels, and Athma A. Pai. "Challenges in identifying mRNA transcript starts and ends from long-read sequencing data." bioRxiv (2023): 2023-07.
Related Services
Full-Length Transcript Sequencing (Iso-Seq)
Nanopore Direct RNA Sequencing
Nanopore Full-Length cDNA Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment