We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Services

circRNA Full-Length Identification and Quantification Service

Full-Length Transcript Sequencing (Iso-Seq)

Nanopore Direct RNA Sequencing

Nanopore Full-Length cDNA Sequencing
Full-Length Transcriptome Profiling Combined with Long- and Short-Read Sequencing

Full-Length Spatial-Temporal Transcriptome Profiling

Single-Cell Full-Length Transcriptome Sequencing
Transcriptome Sequencing: Unveiling BCR/TCR Immune Repertoire
At a glance:
- Innovative Approaches in Immunological Research
- Traditional Methods vs. Novel Transcriptome-Based Analysis
- Analysis Software for BCR/TCR Immune Repertoire
- Case Study
- Summary
Transcriptome sequencing, as a cornerstone of omics technologies in life sciences, produces highly comprehensive and rich gene expression data, representing a vast repository of information. Historically, the focus of data analysis from both bulk RNA sequencing and single-cell or spatial transcriptomics has been centered on deciphering gene expression levels and delving into post-transcriptional modifications, such as alternative splicing, alternative polyadenylation (APA), and RNA editing. Nevertheless, considering the expansive landscape of transcriptome sequencing data, one may wonder if there remain unexplored perspectives and methodologies. The answer is unequivocally affirmative.
Innovative Approaches in Immunological Research
An innovative approach involves utilizing transcriptome sequencing data to explore the B-cell receptor (BCR) and T-cell receptor (TCR) immune repertoires in samples containing immune cells. BCRs and TCRs are hallmark genes of B and T immune cells, respectively, generated through complex recombination mechanisms, resulting in highly diverse clonotypes. These clonotypes encode proteins that play pivotal roles in executing specific humoral and cellular immune functions, making them indispensable for the advancement of immunological research.
Traditional Methods vs. Novel Transcriptome-Based Analysis
Traditionally, the identification and quantification of BCR/TCR clonotypes have relied on the design of specific primers to amplify BCR and TCR sequences, followed by high-throughput sequencing—commonly known as immune repertoire sequencing. However, it is noteworthy that BCRs and TCRs, being protein-coding genes, also produce mRNAs that can be captured through transcriptome sequencing. This discovery opens a new avenue for analyzing BCR/TCR immune repertoire information based on transcriptome sequencing data, highlighting the extensive application potential and innovative value of transcriptome sequencing in immunological studies.
This method not only broadens the scope of transcriptome sequencing applications but also provides an integrated platform for comprehensive immune profiling. By leveraging the vast data generated from transcriptome sequencing, researchers can simultaneously investigate gene expression profiles and immune repertoire diversity. Such a dual approach offers a more holistic understanding of immune responses and cellular dynamics, contributing to the development of more precise and personalized immunotherapies. Moreover, this technique can uncover novel insights into immune mechanisms and disease pathogenesis, thereby driving forward the frontier of immunological research.
Analysis Software for BCR/TCR Immune Repertoire
The expression information of BCRs and TCRs, although present within transcriptomic data, typically represents a minor fraction of the total mRNA. This necessitates the use of specialized software for the extraction and subsequent analysis of immune repertoire data. Among the established tools for immuno-sequencing analysis, MiXCR and TRUST4 are prominent for their capabilities in handling such data.
MiXCR: Precision in Immuno-Repertoire Analysis
MiXCR is a widely utilized tool for the comprehensive analysis of immune repertoires. It is adept at identifying and quantifying the variable, diversity, and joining (VDJ) recombination events characteristic of BCR and TCR sequences. MiXCR employs the rnaseq-cdr3 module, which is specifically designed to process RNA-seq data, enabling the precise extraction of CDR3 (complementarity-determining region 3) sequences. This region is critical for determining the specificity of BCRs and TCRs, as it encompasses the variable region formed by VDJ recombination. The tool's algorithms allow for the accurate identification of these sequences even in the context of low expression levels, which are often observed in bulk RNA-seq datasets.
TRUST4: Versatile Analysis Across Different Sequencing Platforms
TRUST4 (TCR Repertoire Unified System for TCR-seq) extends the capability of immuno-repertoire analysis across various sequencing platforms. It is compatible with bulk RNA-seq, 10x single-cell RNA-seq, and Smart-seq2 datasets, providing a versatile approach to immune repertoire profiling. TRUST4 leverages sophisticated algorithms to reconstruct TCR and BCR sequences, offering insights into the clonality and diversity of immune cells. Its utility in both bulk and single-cell contexts allows researchers to dissect the immune landscape at multiple levels of resolution, from population-wide analyses to single-cell specificity.
The integration of these tools into the analysis pipeline enables a detailed characterization of immune receptor repertoires. By accurately capturing the diversity and specificity of BCR and TCR sequences, researchers can gain profound insights into the adaptive immune response, autoimmunity, and the mechanisms underlying immune recognition and memory.
The deployment of MiXCR and TRUST4 in the analysis of BCR and TCR repertoires from transcriptomic data provides a powerful framework for exploring the complexities of the immune system. These tools facilitate the extraction and characterization of immune receptor sequences, despite their low abundance in RNA-seq datasets. By doing so, they enable a deeper understanding of the molecular underpinnings of immune responses and offer valuable insights into various immunological conditions.
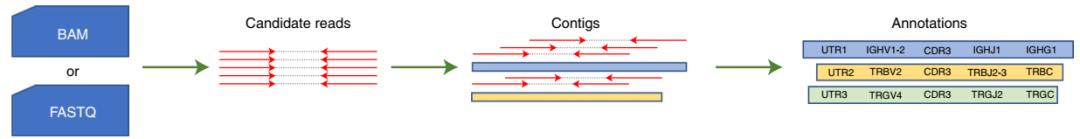 Figure 1. TRUST4's technical principles
Figure 1. TRUST4's technical principles
Service you may intersted in
Case Study
Case Study 1: Identification of BCR/TCR Biomarkers Predictive of Chemotherapy Efficacy in HER2+ Breast Cancer Patients Using RNA-Seq Data
The immunological microenvironment plays a crucial role in determining the treatment outcomes of patients with HER2-positive breast cancer. Identifying immune biomarkers is essential for optimizing chemotherapy efficacy in this patient population. In this retrospective study, RNA-seq data from 518 patients enrolled in the NeoALTTO (N=254) and CALGB 4060 (N=264) phase III clinical trials were analyzed using the MiXCR software. The analysis focused on systematically exploring the complex characteristics of B-cell receptors (BCRs) and T-cell receptors (TCRs) and their associations with treatment outcomes.
The study revealed significant heterogeneity in the number of BCR and TCR clonotypes, clonotype homogeneity, Gini index, and clonal expansion across samples, correlating with variables such as hormone receptor status (HR+/-), PAM50 gene classification, and tumor-infiltrating lymphocyte (TIL) levels. The statistical analysis demonstrated a significant association between BCR clonotype homogeneity and Gini index with event-free survival (EFS) in both cohorts. Furthermore, a predictive model incorporating BCR clonotype characteristics and various clinical parameters was developed, showing high accuracy in forecasting treatment outcomes.
This research underscores the potential of RNA-seq-based immune repertoire analysis in identifying predictive biomarkers for chemotherapy response in HER2+ breast cancer patients. The findings highlight the importance of considering the immune repertoire's diversity and clonality as part of the prognostic assessment in clinical settings.
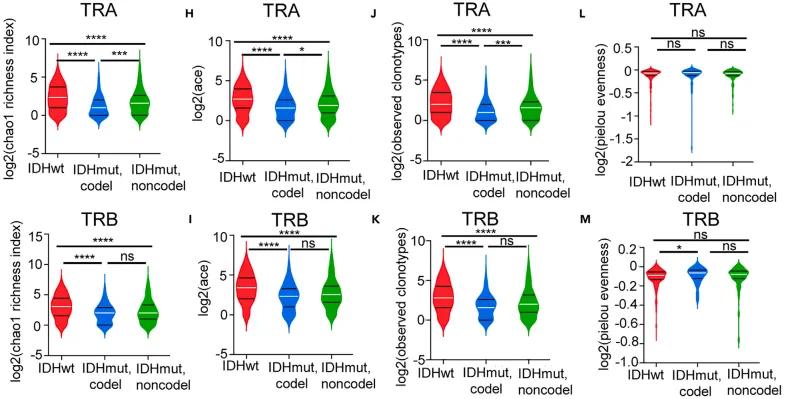 Figure 2. Comparison of TCR clone type chao1 enrichment index in different groups
Figure 2. Comparison of TCR clone type chao1 enrichment index in different groups
Case Study 2: Canine TCR Profiling Based on RNA-seq Data
The study of TCRs in canine models holds significant potential for understanding tumor immunology, given the similarities between canine and human immune systems. However, the existing knowledge of the canine TCR repertoire remains limited, with fewer than 100 TCR clonotypes reported in the literature. This study aims to expand the current understanding by leveraging RNA-seq data from a diverse set of samples, including both normal and tumor tissues.
Data Source and Sample Description
The analysis utilized publicly available RNA-seq data from 1361 samples collected from 1063 dogs, covering a range of tissue types including blood, lymph nodes, and various solid tissues. Among these, 613 samples were derived from tumor tissues. The primary goal was to comprehensively profile the TCR repertoire using state-of-the-art bioinformatics tools.
Bioinformatics Analysis
Two specialized software tools, TRUST4 and MiXCR, were employed to analyze the RNA-seq data. TRUST4 is known for its compatibility with bulk RNA-seq, 10x single-cell RNA-seq, and SMART-seq2 data, making it highly versatile in capturing TCR sequences. MiXCR, another widely used tool, facilitates detailed immune repertoire analysis by accurately identifying and quantifying TCR clonotypes.
The analysis included a comparative evaluation of the results obtained from both software tools, focusing on the usage of TRAV and TRBV genes, identification of novel V genes and their subtypes, and the analysis of complementary determining region 3 (CDR3) length and motif conservation. Further, clonotype expansion and diversity metrics were assessed, with particular attention to identifying disease-specific clonotypes.
Results and Discussion
Comparative Analysis of TRUST4 and MiXCR Outputs
The results demonstrated significant overlaps as well as unique findings between TRUST4 and MiXCR outputs. TRUST4 provided more comprehensive coverage of TRAV and TRBV genes, identifying several novel V gene subtypes that were previously unreported in canine TCR studies. The CDR3 region analysis revealed conserved motifs indicative of shared antigenic specificities, with TRUST4 showing a slightly higher sensitivity in detecting low-frequency clonotypes.
Clonotype Diversity and Expansion
The study observed substantial clonal expansion in tumor samples, characterized by a reduction in TCR diversity. This finding is consistent with the immune response dynamics observed in other species, where clonal expansion is often a marker of an active immune response against malignancies. The Gini index and clonality metrics indicated a more restricted repertoire in tumor tissues compared to normal samples.
Cross-Species Comparison
A cross-species comparison of β-CDR3 sequences between dogs, humans, and mice revealed both conserved and species-specific features. This comparative analysis highlighted the potential for canines to serve as a relevant model for studying human TCR biology, particularly in the context of cancer immunotherapy.
Conclusion
This study significantly expands the current knowledge of the canine TCR repertoire, providing a comprehensive dataset that includes novel gene segments and detailed clonotype diversity metrics. The findings underscore the utility of advanced bioinformatics tools like TRUST4 and MiXCR in immunogenomics research. The identification of disease-specific clonotypes offers promising avenues for future studies aimed at developing canine-specific immunotherapies and improving translational research applications in humans.
Case Study 3: Spatial Transcriptomics-Based Analysis of BCR Repertoires in Tumor Tertiary Lymphoid Structures
Tertiary lymphoid structures (TLS) within tumors are often associated with favorable responses to immunotherapy and positive prognostic outcomes. These ectopic lymphoid formations resemble secondary lymphoid organs and are characterized by the presence of various immune cell subsets, including B cells. The investigation of B-cell responses within TLS can provide critical insights into the tumor-immune microenvironment and potentially unveil biomarkers for therapeutic interventions. This study employs spatial transcriptomics to analyze B-cell receptor (BCR) repertoires within TLS in renal cell carcinoma tissues.
Sample Collection and Sequencing
Fresh frozen renal cell carcinoma tissues were subjected to spatial transcriptomics using a high-depth sequencing approach, achieving a coverage of 150,000 reads per spot. This high-resolution technique enabled the precise localization of immune cells within the tumor microenvironment and provided comprehensive coverage of the CDR3 regions of both the immunoglobulin heavy (IGH) and light (IGL) chains.
Data Analysis
The BCR repertoire was analyzed using the MiXCR software, which facilitates the identification and quantification of BCR clonotypes from RNA-seq data. Additionally, 5'-bulk RNA sequencing was performed to corroborate the findings from the spatial transcriptomic data, ensuring a robust analysis of BCR diversity and clonal expansion.
Results and Discussion
B-Cell Localization and Maturation in TLS
The spatial transcriptomics data revealed a significant enrichment of B cells within the TLS regions of the tumor. Notably, a maturation gradient was observed, indicating a transition from naïve B cells to plasma cells. This finding suggests active local differentiation of B cells within the tumor microenvironment, potentially driven by antigenic stimulation.
BCR Repertoire Characteristics
The BCR analysis identified distinct somatic hypermutations (SHM) and clonal expansions specific to the TLS regions. These somatic mutations are indicative of antigen-driven selection processes occurring within the TLS, akin to germinal center reactions observed in secondary lymphoid organs. The presence of TLS-specific SHM and clonal expansions underscores the role of these structures in orchestrating localized immune responses against the tumor.
Implications for Immunotherapy
The identification of unique BCR clonotypes and SHM within TLS provides valuable insights into the adaptive immune landscape of renal cell carcinoma. These findings suggest that TLS may serve as critical sites for the generation and maintenance of anti-tumor immunity. Furthermore, the TLS-specific BCR repertoires could serve as potential biomarkers for predicting responses to immunotherapy and tailoring personalized therapeutic strategies.
Conclusion
This study highlights the utility of spatial transcriptomics in elucidating the complex immune architecture within tumors. The detailed characterization of BCR repertoires in TLS not only enhances our understanding of the tumor immune microenvironment but also opens avenues for developing novel immunotherapeutic approaches. The findings underscore the potential of TLS as therapeutic targets and prognostic indicators in renal cell carcinoma.
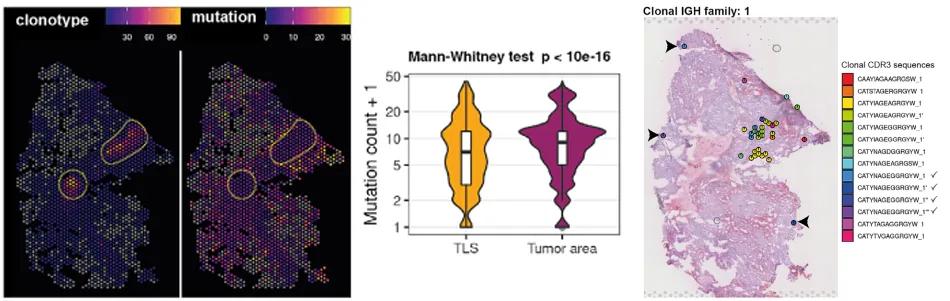 Figure 3. Spatial distribution and mutation frequency of BCR clonotypes in the TLS region
Figure 3. Spatial distribution and mutation frequency of BCR clonotypes in the TLS region
Summary
With the extensive publication of RNA-seq data for various tumors and other diseases, coupled with the development and maturation of relevant software such as MiXCR and TRUST4, the extraction of BCR and TCR information from RNA-seq data has become highly feasible. This approach offers several advantages over immune repertoire sequencing, which tends to be more costly and less available in public datasets. Transcriptome-based immune repertoire analysis facilitates large-scale cohort studies, enabling the discovery of significant new findings across substantial sample sizes.
Furthermore, this strategy presents a viable alternative for studying other mammalian species beyond humans and mice. It mitigates the challenges associated with designing species-specific primers for BCR and TCR, thus broadening the applicability of immune repertoire research to a wider range of species. This expanded scope is particularly valuable for understanding immune responses in non-model organisms, potentially leading to novel insights into comparative immunology and the evolution of adaptive immunity.
In summary, the integration of transcriptome sequencing and immune repertoire analysis presents a transformative opportunity in immunology. It fosters the discovery of novel biomarkers, enhances our understanding of immune cell functionality, and ultimately contributes to the development of innovative therapeutic strategies. As this approach continues to evolve, it promises to redefine the landscape of immune profiling and expand the boundaries of transcriptome research.
References
- Bolotin D A, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nature methods, 2015, 12(5): 380-381.
- Song L, Cohen D, Ouyang Z, et al. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nature methods, 2021, 18(6): 627-630.
- Rediti M, Fernandez-Martinez A, Venet D, et al. Immunological and clinicopathological features predict HER2-positive breast cancer prognosis in the neoadjuvant NeoALTTO and CALGB 40601 randomized trials. Nature communications, 2023, 14(1): 7053.
- Zhang M, Ho K L, Zhao S. Initial Characterization of Canine T-cell Receptor Repertoires Using RNA-seq Data from Different Diseases and Tissues. bioRxiv, 2024: 2024.02. 14.580390.
- Meylan M, Petitprez F, Becht E, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity, 2022, 55(3): 527-541. e5.
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment