We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Pan-Genome Analysis: Overview, Workflow, Application and Recent Advances
At a glance:
- What is pan-genome?
- How to build a pan-genome?
- What is the difference between pan and core genome?
- Application of pan-genome
- Advances in pan-genome assembly technologies
- PacBio HiFi sequencing, a good solution for pan-genome construction
What is pan-genome?
A pan-genome is the sum of all genomic information within a species. With the development of genomic technology, researchers have found that a single reference genome can no longer meet the needs of genomic data analysis, and more and more species, including the human genome, are choosing to construct a pan-genome instead of a single reference genome.
Pan-genomes reflect structural variation (SV) and polymorphisms in the genome, allowing in-depth comparisons of variation at the species level or at higher taxonomic levels. Pan-genomes have potential applications in crop improvement, evolution and biodiversity research. To fully exploit the value of pan-genomes, a broader range of information such as phenotypic, environmental and expression data needs to be integrated to provide insight into the role of variable regions in the genome.
How to build a pan-genome?
There is extensive genomic diversity within species, and a pan-genome need to capture this diversity while removing redundancies to generate an integrated single genome.
Map to pan, which starts with de novo assembly, and matches the sequences of each individual assembled to the reference genome to find the unmatched sequences, then finds all the unmatched sequences and builds the pan-genome, or iterative mapping and assembly methods
Iterative assembly starts with a single reference genome and then complements it with non-redundant sequences from other individuals or the iterative mapping and assembly method starts from a single reference genome and then complements it with non-redundant sequences from other individuals to build a pan-genome.
De novo assembly requires the individual genomes to be assembled separately, followed by whole genome comparison.
What is the difference between pan and core genome?
Pan-genomic analysis clusters gene sets by co-occurrence in each individual and is usually divided into three categories: Core gene, genes present in all plant and animal strains; dispensable gene, genes present in one or more plant and animal strains; private, genes present in only one strain. The core part is present in all individuals, while the dispensable part is present in only one individual.
Application of pan-genome
Pan-genomic analysis helps to understand the characteristics of species, while the complex genomic variation provided by pan-genome mapping helps to resolve the diversity of crop phenotypes and agronomic traits.
- Selection of different subspecies for pan-genome sequencing allows the study of important biological questions such as the origin and evolution of species
- Selecting germplasm resources with different characteristics, such as wild species and cultivated species, for pangenome sequencing can uncover genetic resources related to important traits and provide guidance for scientific breeding
- Selecting germplasm resources of different ecogeographic types for pangenome sequencing can carry out popular scientific questions such as adaptive evolution of species and invasiveness of exotic species
- The use of crop pangenome advancement QTL mapping and GWAS can be used to identify genomic regions associated with desired phenotypes
- The use of pangenomes to advance genomic prediction. With SNPs as predictors, important agronomic traits such as grain yield, grain moisture, grain quality, biomass traits, and stem and root collapse can be predicted with reasonable accuracy
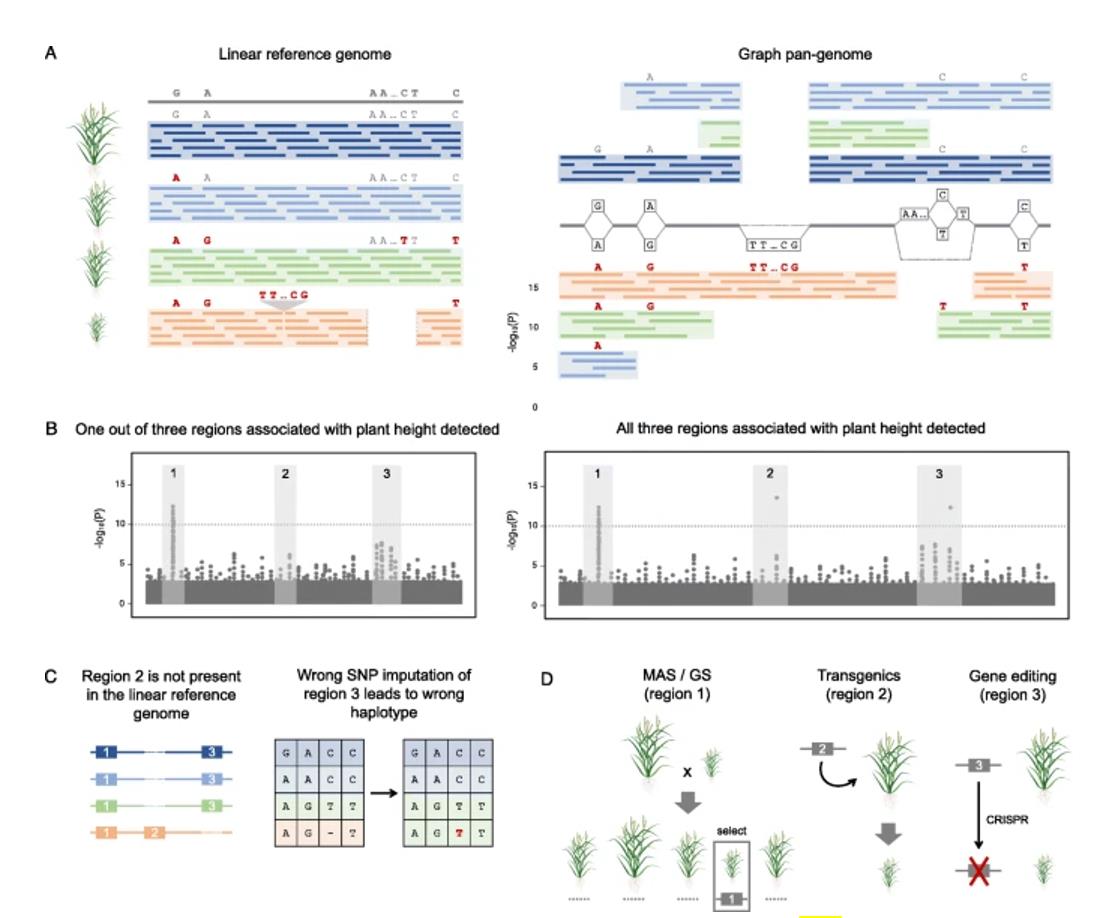 Application of pan-genome in crop improvement (Della Coletta R et al., 2021)
Application of pan-genome in crop improvement (Della Coletta R et al., 2021)
Advances in pan-genome assembly technologies
The reduced cost of Illumina sequencing and improvements in assembly algorithms have facilitated the use of low-cost short-read data (e.g., maize genome, rice genome, soybean genome). While this approach has generated highly complete and contiguous assemblies of low-copy gene regions, the more repetitive, TE-rich regions of the genome have proven difficult to assemble with short reads, resulting in large gaps and partial assemblies in these regions. Recently, the maturation of long-read sequencing technologies, especially PacBio HiFi Sequencing, has facilitated more contiguous and complete assemblies of crop genomes and, in some cases, long-read length-based assemblies within a single species. Advances in PacBio pangenome sequencing technologies are described below.
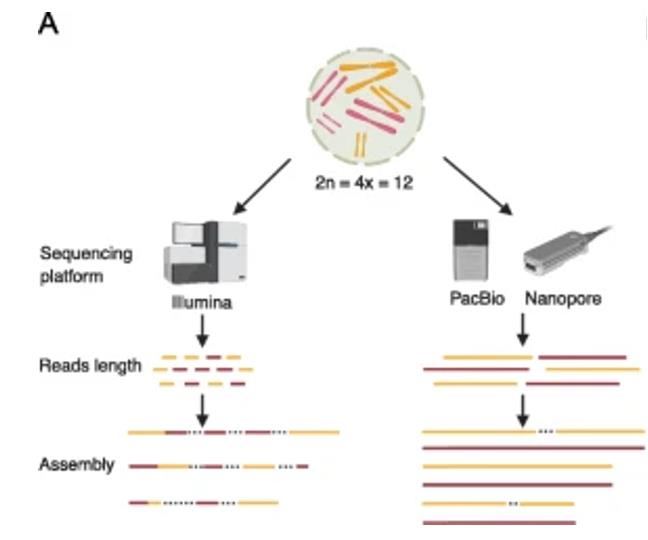 Impact of sequencing technology on polyploid assembly (Della Coletta R et al., 2021)
Impact of sequencing technology on polyploid assembly (Della Coletta R et al., 2021)
PacBio HiFi sequencing, a good solution for pan-genome construction
Nowadays, pan-genome construction generally uses three-generation long read-length sequencing to assemble multiple samples of a population from scratch. The two technology platforms now commonly used for triple sequencing are PacBio's HiFi sequencing and ONT's Nanopore sequencing, of which HiFi sequencing takes into account long read length and ultra-high accuracy, and is extremely suitable for sequencing genomic de novo assembly.
- HiFi reads are more accurate
The higher accuracy of HiFi reads allows the assembly algorithm to extend contigs to flanking mitotic regions with high confidence through more repeat assemblies at shorter read lengths, enhancing the integrity of the mitotic and telomeric regions
- Simplify the complexity of polyploid genome assembly
High-quality genome assembly in polyploid species has been difficult to achieve due to the inclusion of multiple closely related subgenomes and the associated challenges in distinguishing homologous motifs and creating non-mosaic subgenomic scaffolds. Long-read sequencing with low error rates (e.g., PacBio HiFi read length) has enabled high-quality polyploid genome assembly, with recent assemblies containing fewer gaps and resolved homologous scaffolds. As polyploid pangenomes of more species are revealed, more novel structural variants and markers are likely to be observed.
References
- Della Coletta R, Qiu Y, Ou S, et al. How the pan-genome is changing crop genomics and improvement. Genome biology, 2021, 22(1): 1-19.
- Leonard, Alexander S., et al. Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies. Nature communications 13.1 (2022): 3012.
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment