
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Mitochondrial DNA Profiling:Methods and Advanced Analyses
At a glance:
- 1. Genome Assembly and Annotation
- 2. Genome Composition Analysis
- 3. Sequence Submission and Data Visualization
- 4. Advanced Analyses
- 5. Conclusion
Mitochondrial DNA plays a crucial role in molecular evolution, biosystematics, and population genetics. Due to its unique genetic characteristics-small-scale genomes, maternal inheritance, and high mutation rates-mitochondrial genomes have become essential tools for studying species evolution, taxonomy, and ecology.This article systematically outlines the fundamental methods and steps for mitochondrial genome analysis, delving into advanced analytical techniques to provide practical references for related studies. Through these approaches, researchers can more precisely analyze genomic variations, trace evolutionary trajectories, and understand biological adaptation mechanisms.
1. Genome Assembly and Annotation
Accurate assembly and precise annotation of mitochondrial genomes are critical for downstream analyses. Below, we discuss commonly used assembly software and gene annotation tools.
1.1 Assembly Software
Mitochondrial genome assembly relies primarily on data generated by high-throughput sequencing. Frequently used tools include:
Staden Package: A sophisticated molecular sequence analysis platform specifically engineered for comprehensive genome reconstruction. This toolkit excels in mitochondrial and small genomic assemblies, providing automated and manually configurable processing capabilities. It demonstrates exceptional versatility across multiple sequencing technological platforms, particularly effective for plant and animal mitochondrial genomic investigations.
Integrated Genome Assembly Solutions: ContigExpress, DNAMAN, and DNASTAR represent advanced computational platforms designed for high-precision genomic reconstruction. DNASTAR distinguishes itself through comprehensive annotation features, making it particularly valuable for intermediate-scale research methodologies.
Specialized Sequence Manipulation Applications:Sequencher and BioEdit emerge as sophisticated sequence editing instruments, optimized for intricate sample processing requiring extensive manual refinement and nuanced genomic interpretation.
1.2 Gene Annotation
Gene annotation is crucial for understanding the functional aspects of mitochondrial genomes, including protein-coding genes, rRNA, tRNA genes, and non-coding regions (e.g., A+T-rich regions). Common tools include:
- DOGMA: An online tool specifically for mitochondrial gene annotation, capable of identifying most functional genes.
- MITOS: Provides an automated annotation pipeline for mitochondrial genomes across different species.
- tRNAscan-SE and ARWEN: Highly accurate tools for identifying tRNA genes in mitochondrial genomes, supporting multiple species.
- BLAST and MiTFi: Useful for aligning and predicting the functions of specific genes, particularly for correcting errors in automated annotations.
2. Genome Composition Analysis
Genome composition analysis is essential for exploring mitochondrial genome characteristics. Tools such as MEGA5 are widely used for analyzing nucleotide composition, gene content, and codon usage bias.
Nucleotide Composition Analysis: Includes proportions of A, T, G, and C, as well as GC content, revealing evolutionary traits of the mitochondrial genome.
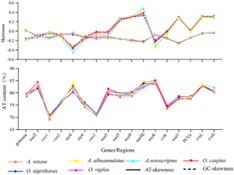 Fig.1. AT/GC contents and AT/GC-skews of the investigated mitogenomes. (Ma, X. X.,et.al,2022)
Fig.1. AT/GC contents and AT/GC-skews of the investigated mitogenomes. (Ma, X. X.,et.al,2022)
Gene Content Analysis: Provides information on gene types and quantities, forming the basis for interspecies comparisons.
3. Sequence Submission and Data Visualization
The sharing and visualization of mitochondrial genome data foster scientific collaboration and communication.
3.1 Sequence Submission
Sequin: Developed by NCBI, this tool is designed for submitting mitochondrial genome sequences to GenBank. The submission process involves sequence organization, metadata completion, and annotation verification.
3.2 Data Visualization
Tools such as CGview, MTviz, and OGDRAW generate high-quality circular genome maps, illustrating gene distribution and structural features of the genome.
4. Advanced Analyses
Advanced analytical techniques broaden the applications of mitochondrial genome research, including phylogenetics, evolutionary analysis, and functional predictions.
4.1 Synteny Analysis
Collinearity refers to the genetic linkage of genes, a phenomenon where homologous genes are arranged in the same order on the chromosomes of different species. The degree of collinearity between two species can serve as a measure of their evolutionary distance, providing insight into their evolutionary relationships. Analyzing local collinear blocks between genomes for phenomena such as similarity, rearrangements, and inversions can help elucidate events in species evolution. Commonly used analysis tools include MUMmer, LASTZ, and others.
4.2 Phylogenetic Tree Construction
A phylogenetic tree, also known as a phylogeny, is a diagram that illustrates the evolutionary relationships among species, typically represented in a tree-like branching structure. It can be used to describe the evolutionary connections between different species. Phylogenetic tree analysis allows the identification of evolutionary relationships between species, helps understand the relationship between ancestral sequences and their descendants, and can also estimate the divergence time between species that share a common ancestor.
Organelle genomes are very conserved and are commonly used to construct phylogenetic evolutionary trees to study the species classification and evolutionary status of plants and animals. Ma, X. X.,et.al used 13 PCGs from 30 taxa via two analytical approaches to construct phylogenetic tree.
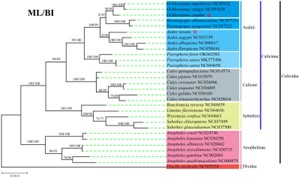 Fig. 2 Phylogenetic analysis of 13 PCG amino acids. (Ma, X. X.,et.al,2022)
Fig. 2 Phylogenetic analysis of 13 PCG amino acids. (Ma, X. X.,et.al,2022)
4.3 Selective Pressure Analysis
Selective pressure refers to the external pressures exerted on a species during its evolutionary process, driving the species to adapt to its natural environment. In genetics, ω = Ka/Ks or dN/dS represents the ratio between nonsynonymous mutations (Ka) and synonymous mutations (Ks). Synonymous mutations are generally considered to be neutral to natural selection, whereas nonsynonymous mutations are subject to selective forces. Typically, ω > 1 indicates positive selection, meaning that beneficial mutations are being actively selected; ω = 1 suggests neutral evolution, with no selective pressure; and 0 < ω < 1 indicates purifying selection, where harmful mutations are being removed. A smaller ω value implies stronger negative selection, resulting in a more conserved amino acid sequence. This ratio is commonly used to assess whether a gene has undergone positive selection (beneficial mutations) or purifying selection (removal of harmful mutations).
4.4 Organelle and Nuclear Genome Exchange Analysis
By comparing mitochondrial genome fragments with nuclear genome fragments, patterns of structural variations and insertions/deletions (InDels) can be revealed. Fragment exchange between mitochondrial and chloroplast genomes is a common phenomenon in higher plants, with approximately 5%-10% of mitochondrial genome sequences in different species showing homology to chloroplast genome sequences. This analysis is of significant importance for exploring the mechanisms of horizontal gene transfer in chloroplast genomes and understanding its role in plant evolution.
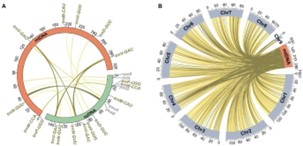 Fig3. Analysis of nucleotide polymorphisms in mitochondrial genome. (Jiang, M,et.al,2023)
Fig3. Analysis of nucleotide polymorphisms in mitochondrial genome. (Jiang, M,et.al,2023)
4.5 Genome Polymorphism Analysis
Nucleotide polymorphism (Pi) is a parameter used to measure the level of genetic diversity within a specific population. It is defined as the average nucleotide differences observed between two randomly selected DNA sequences at each nucleotide position within the population. Pi serves as an indicator of sequence variation among species, with highly variable regions offering potential molecular markers for population genetics studies.
4.6 Core and Specific Gene Analysis
Homologous genes present in all samples are referred to as "core genes." After excluding core genes, the remaining ones are categorized as "dispensable genes," while "specific genes" are those uniquely present in a particular sample. Core and specific genes are likely associated with the commonalities and distinctive characteristics of the samples, making them valuable for investigating functional differences between samples.
4.7 Codon Usage Bias Analysis
The relative probability of a specific codon among the synonymous codons that encode the corresponding amino acid can reflect the degree of codon preference. The codon preference value is obtained by calculating the Relative Synonymous Codon Usage (RSCU). Studying codon usage patterns is important for understanding evolutionary pressures on a species and for further genetic research.
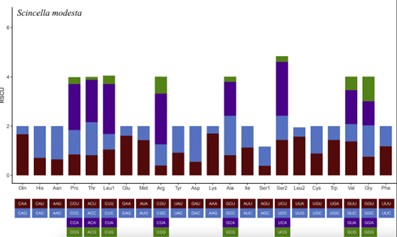 Fig4. Codon bias analysis. (Chen, L,et.al,2021)
Fig4. Codon bias analysis. (Chen, L,et.al,2021)
4.8 Simple Sequence Repeat (SSR) Analysis
Simple Sequence Repeats (SSRs), also known as Microsatellite Sequences (MS), are DNA fragments composed of repeated units of 1-6 nucleotides. SSRs are abundant, highly polymorphic, uniformly distributed across the genome, exhibit codominant inheritance, and are easy to detect. Due to these characteristics, SSRs are widely used as second-generation molecular markers in various fields such as genetic mapping, target gene localization, genetic diversity studies, molecular-assisted breeding, and germplasm resource identification.
4.9 tRNA Secondary Structure Analysis
tRNA is a key molecule in decoding genetic codes in mRNAs and protein synthesis. Typically consisting of 76 nucleotides, tRNA adopts a secondary structure resembling a cloverleaf, comprising three stem-loop regions: the D loop (which contains a dihydrouridine loop), the anticodon loop, and the T loop (which includes thymidine, pseudouridine, and either cytidine or TΨC loop).
4.10 RNA Editing Analysis
Mitochondrial gene expression requires complex post-transcriptional processing, including RNA C-U editing, intron splicing, 5' and 3' end maturation, and RNA stability. RNA editing is widespread in plant organelles (mitochondria and chloroplasts) and is essential for plant growth and development. RNA editing can be broadly defined as any site-specific change in the RNA sequence that may differ from the template during replication.
5. Conclusion
Comprehensive mitochondrial genomic investigations represent a multifaceted scientific approach bridging molecular sequencing, structural characterization, and advanced functional analyses. The methodological framework encompasses critical research strategies including genomic reconstruction, precise annotation, compositional evaluation, systematic data archiving, and sophisticated visualization techniques.
Advanced analytical methodologies extend beyond fundamental approaches, incorporating specialized investigations such as comparative syntenic mapping, phylogenetic reconstruction, and evolutionary selective pressure assessments. These integrated research techniques provide researchers with sophisticated instruments for deciphering intricate genetic mechanisms, evolutionary dynamics, and functional complexities inherent in mitochondrial genomic structures.
Technological innovations in high-throughput sequencing and computational bioinformatics are progressively expanding research capabilities. Emerging methodological paradigms are positioned to generate transformative insights across molecular biology, evolutionary genetics, and related biomedical disciplines, promising unprecedented understanding of mitochondrial genomic architectures and their broader biological significance.
References
- Ma, X. X.,et.al. (2022). First description of the mitogenome and phylogeny:Aedes vexansand Ochlerotatus caspius of the Tribe Aedini (Diptera: Culicidae). Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases, 102, 105311. https://doi.org/10.1016/j.meegid.2022.105311
- Jiang, M,et.al (2023). Characterisation of the complete mitochondrial genome of Taraxacum mongolicum revealed five repeat-mediated recombinations. Plant cell reports, 42(4), 775–789. https://doi.org/10.1007/s00299-023-02994-y
- Chen, L,et.al.(2021). Characterization of the complete mitochondrial genome of the many-lined sun skink (Eutropis multifasciata) and comparison with other Scincomorpha species. Genomics, 113(4), 2526–2536. https://doi.org/10.1016/j.ygeno.2021.05.030
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment