We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Transcriptome Analysis of Human Breast Cancer Cell Lines MCF-7
At a glance:
- Overview
- Why Choose MCF-7
- Single-cell Transcriptome Analysis of MCF-7
- Long-read Transcriptome Analysis of MCF-7
Overview
It is well known that cancer is caused by the gradual accumulation of genomic and epigenomic alterations leading to dysregulated cell growth. Chromosomal instability (a hallmark of cancer) greatly contributes to the process of oncogenesis and subsequent cancer progression, with continuous mutations in the genome and epigenome of each cancer cell leading to a diverse transcriptome in cancerous tissues. This diversity leads to differences in drug response between individual cancer cells and results in poor prognosis. Therefore, it is important to reveal the diversity of the transcriptome profiles of individual cancer cells in order to further understand how diverse cancer cells interact with each other, enabling cancer cell populations to evade immune surveillance and respond poorly to drug therapy.
Thanks to advances in next-generation sequencing (NGS) technology, it is possible to analyze cancer genomes and associated transcriptomes at the single-cell level. RNA sequencing (RNA-Seq) is widely used to study the human breast cancer cell line MCF-7. Although advances in RNA sequencing methods have accelerated our understanding of the human transcriptome, the discovery of complete mRNA isoforms remains a challenge because short reads require complex assembly algorithms to infer the continuity of full-length transcripts. This inability to resolve full-length isoforms hampers researchers' ability to identify key isoform changes and fusion transcripts associated with human disease. With PacBio SMRT Sequencing Technology, it is now possible to sequence full-length transcript isoforms up to 10 kb in length. SMRT sequencing generates reads from independent observations of single molecules; assembly is not required if the reads span the entire length of the transcript. SMRT sequencing technology gives researchers the ability to new opportunities to study the complex splicing events that occur in cancer cells.
Why Choose MCF-7
MCF-7 is one of the most widely used metastatic breast cancer cell lines. It retains several features of differentiated mammary epithelium. On the other hand, breast cancer cells such as HER2-positive and TNBC (triple-negative breast cancer) have undergone an intensive selection process that abolishes the expression of some key genes, which may further affect the expression of many other genes, thereby eliminating some key genes. gene expression. Making the transcriptome more isoform-specific. MCF-7 represents a more prevalent form of breast cancer and is more likely to express both consistently and discordantly expressed genes. These features make MCF-7 an ideal candidate for transcriptome studies, as deciphering its inherent heterogeneity can provide insight into its clinical behavior and response to therapy.
Single-cell Transcriptome Analysis of MCF-7
Traditional sequencing methods can capture average gene expression across populations of cells, but these methods can mask key variations that exist within individual cells. In recent years, microarrays, high-throughput cDNA sequencing, and RNA-seq have become very useful tools for studying the transcriptome. Single-cell transcriptome analysis provides a fine-grained view of the transcriptome landscape of each cell. The technology amplifies the unique transcriptome signature of each cell, enabling identification of rare cell populations, elucidation of cellular pathways, and discovery of novel therapeutic targets.
Data obtained using MCF-7 single-cell transcriptome profiling could revolutionize the treatment landscape for breast cancer. Potential applications include:
- Targeted drug development: Identifying consistently and discordantly expressed genes can guide the development of drugs targeting these specific genes, providing more targeted treatments.
- Personalized therapy: By understanding the unique transcriptomic signature of each patient's tumor cells, clinicians can prescribe more effective treatments and reduce the trial-and-error approach common in cancer treatment.
- Overcoming drug resistance: Gaining insight into inconsistent gene expression patterns could help develop strategies to overcome drug resistance, a major obstacle to cancer treatment.
Long-read Transcriptome Analysis of MCF-7
SMRT sequencing technology can generate a deep dataset of RNA full-length cDNA sequencing of the human breast cancer cell line MCF-7, helping researchers gain insight into the complexity of the MCF-7 transcriptome. Long-read sequencing has the following advantages in the transcriptome analysis of the human breast cancer cell line MCF-7:
Unparalleled Resolution
Unlike short-read technologies, long-read sequencing can read entire RNA molecules without complex assembly algorithms. This capability ensures that the continuity of full-length transcripts is preserved, giving researchers an unimpeded view of isoform diversity.
Fusion Transcript Discovery
The MCF-7 cell line, in particular, demonstrated the power of long-read sequencing to discover fusion transcripts. For example, the BCAS3-BRCA4 fusion presented multiple fusion transcripts, some of which were de novo. These findings highlight potential therapeutic targets and diagnostic markers.
Isoform Complexity
Sequencing of the MCF-7 cell line using the Clontech SMARTer® cDNA Preparation Kit revealed a surprising number of unique full-length transcript sequences. These sequences were mapped back to the human genome with exceptional accuracy, reflecting the transcriptional diversity of the cell lines. With an average of 3 isoforms per site, the depth of splicing events becomes apparent.
Verify Long-read Data
Validation of transcript data is critical. For the MCF-7 dataset, the predicted peptides were compared to a previously cataloged mass spectrometry dataset of the same cell line. The vast majority of peptides mapped precisely to PacBio predicted open reading frames, confirming the translational potential of these transcripts. This validation step enhances the precision and usefulness of long-read sequencing data.
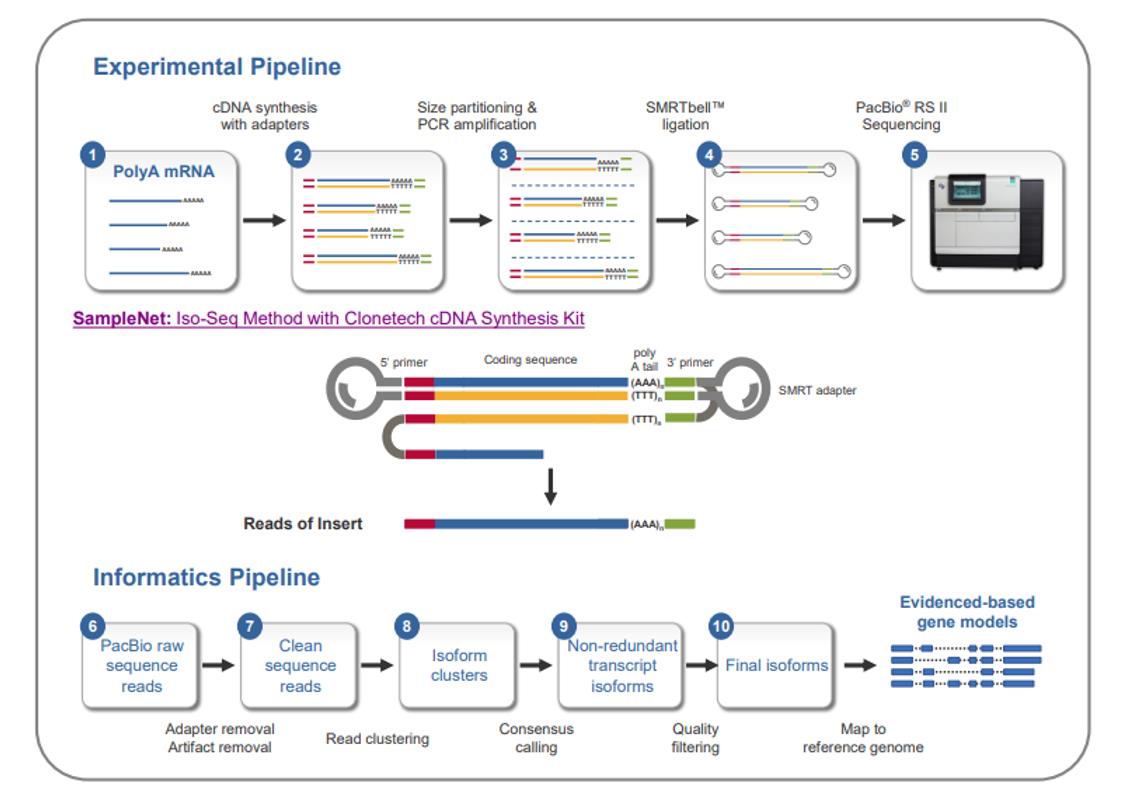 The Iso-Seq Method sample prep and analysis workflow. (Tseng et al., 2015)
The Iso-Seq Method sample prep and analysis workflow. (Tseng et al., 2015)
References
- Chiang, Yih-Shien, et al. "Single cell transcriptome analysis upon MCF-7 breast cancer." Genome Biology. 11.1 (2010): 1-1.
- Tseng, Elizabeth, et al. "Full-length Isoform Sequencing of the Human MCF-7 Cell Line using PacBio® Long Reads." peptides. 50.41,699 (2015): 50-594.
Related Services
PacBio SMRT Sequencing Technology
Single-Cell Full-Length Transcriptome Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment