We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Repeat Expansions Analysis
At a glance:
- Overview
- What are Short Tandem Repeats
- Repeat Expansion Diseases
- Methods for Characterizing Short Tandem Repeats
- Straglr: A New Software Tool for Repeat Expansion
Overview
Repeat expansion, a structural variation in which the number of tandem DNA sequences doubles, has long been associated with many genetic and neurological disorders. However, despite the well-documented contribution of tandem repeats (TRs) to genetic variation, TRs remain poorly understood and their impact on phenotypes and disease may be underestimated. This is largely attributable to the repetitive nature, high GC content, and length of these extensions, which make them difficult to amplify by PCR. Furthermore, repeat extensions are often over 10 kb in length, and many cannot be spanned by short reads, presenting a major challenge to their accurate computational resolution.
The critical role of TR in disease pathogenesis requires the adoption of advanced techniques for accurate and detailed detection. Two main approaches that stand out in the context of analyzing these extensions are short-read sequencing and long-read sequencing. Long-read sequencing reads can span repeat extensions from start to finish in a single read without the need for PCR, enabling accurate determination of the size of extended repeats.
What are Short Tandem Repeats
Short tandem repeats (STRs) are DNA sequences consisting of 2-6 base pair (bps) units that repeat in tandem and in the same orientation. These genomic segments play an integral role in the human genome, accounting for about 3 percent. Their high degree of polymorphism means that multiple variants can exist in a population, which has applications in fields such as forensic genetics, enabling individual identification. Several human diseases are caused by STR amplification, where STR lengths above a certain threshold are considered pathogenic. Threshold length varies by STR locus and disease, and longer repeats may lead to more severe and earlier clinical symptoms in patients with certain repeat expansion disorders. Furthermore, repeat size may also be strongly associated with heterogeneity in phenotypic expression, even among patients with the same disease. Trinucleotides are repeating units or repeat motifs that have hitherto been found primarily in disease, but more recently longer and more complex expansions of tandem repeats (TRs), some of which are >6 bp in size (termed Tandem Repeats). variable number tandem repeat) repeat (VNTR) has been associated with neurological disorders.
Repeat Expansion Diseases
Fragile X Syndrome
Perhaps one of the best-studied disorders caused by STR amplification is fragile X syndrome, the leading genetic cause of intellectual disability and autism. It is caused by an expansion of the CGG repeat located in the 5' untranslated region of the FMR1 gene on the X chromosome. While most unaffected individuals have 6 to 54 repeats, affected males often have more than 200 repeats.
Huntington's Disease
Huntington's disease is another classic example, affecting about 5 in 100,000 Caucasians. This neurodegenerative disorder is due to an expansion of the CAG repeat sequence in the coding sequence of the huntingtin gene. The threshold for disease onset was 35 repeat motifs, with unaffected individuals possessing fewer repeat motifs.
Other Disorders Associated with Repeat Expansion
Many diseases are caused by STR amplification. A prime example includes the hexameric GGGGCC expansion in the C9orf72 gene, which causes amyotrophic lateral sclerosis and frontotemporal dementia. This expansion significantly contributed to the highest genetic risk for both diseases compared with any other single locus. It must also be emphasized that Fuchs' endothelial corneal dystrophy arises from a CTG amplification of the TCF4 gene.
Furthermore, recent studies revealed a unique pentameric repeat in the SAMD12 gene that leads to FAME1. Interestingly, this specific duplication is not present in the general population, highlighting the subtle ways in which STRs affect health.
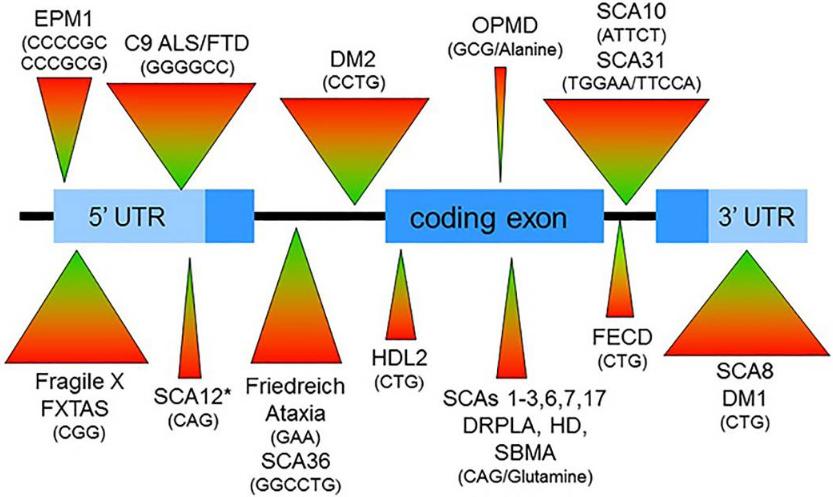 Schematic of gene showing repeat expansions that cause neurological diseases(Paulson et al., 2018)
Schematic of gene showing repeat expansions that cause neurological diseases(Paulson et al., 2018)
Methods for Characterizing Short Tandem Repeats
Polymerase Chain Reaction (PCR) or Southern blot
For many repeat expansion disorders, PCR-based repeat length screening is straightforward, sensitive, specific, and inexpensive. Specific candidate genetic tests are readily available and cost-effective in the right clinical setting. However, for some repeat expansion diseases, especially those with very large and complex expansions or possible interruption of pure repeats in some individuals, Southern blot hybridization analysis complicates the interpretation of results. In conclusion, both methods can only be used to analyze one or a few target STR loci at a time.
This method involves sequencing DNA fragments of shorter length (typically 50 to 500 base pairs). Technologies such as Illumina sequencing primarily employ this approach.
Advantages:
- High precision: Short-read sequencing platforms such as Illumina have high precision and low error rates, ensuring accurate base pair reads.
- Throughput: Given the parallel processing of millions of sequences, short-read platforms provide high throughput, enabling the analysis of large datasets in a relatively short period of time.
Limitations:
- Length limitations: Due to their inherent nature, short-read sequencing has difficulty fully spanning large repeat expansions, especially those exceeding a few kilobases.
- Computational complexity: Resolving large tandem repeats requires complex computational tools, often resulting in inaccurate or unresolvable sequences.
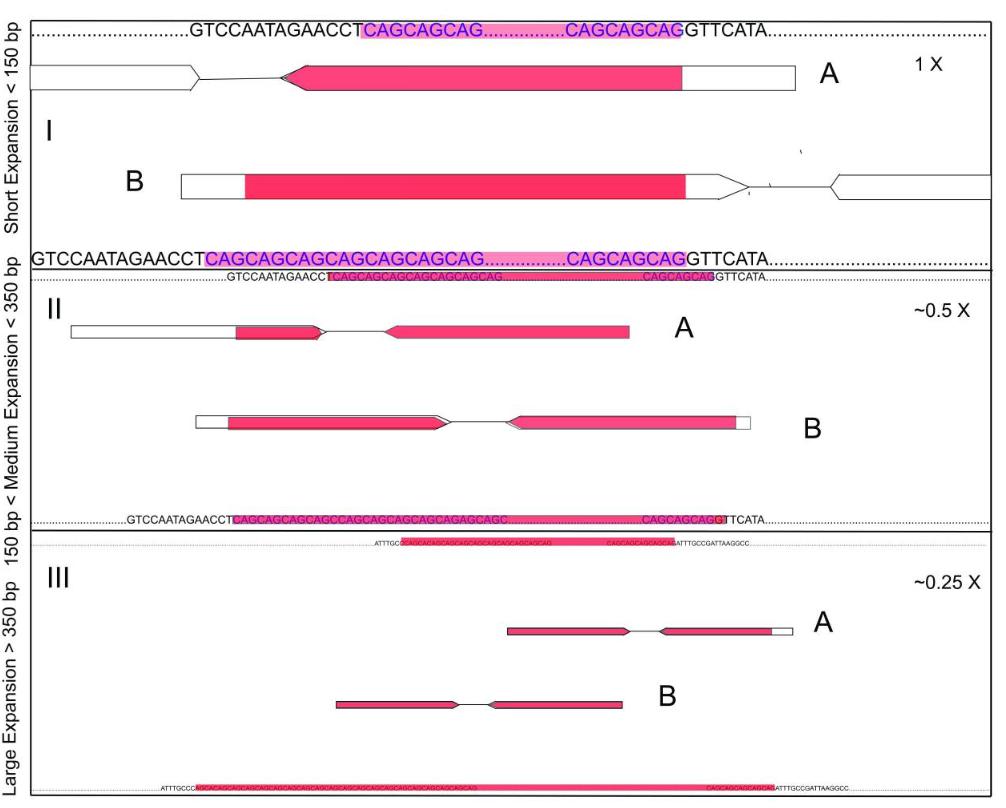 Detecting repeat expansions with short-read sequencing data. (Bahlo et al., 2018)
Detecting repeat expansions with short-read sequencing data. (Bahlo et al., 2018)
Long-read sequencing technologies, such as PacBio and Nanopore sequencing, can read significantly longer DNA fragments, typically tens of thousands of base pairs. The technology can sequence entire repeat expansions, regardless of their size.
Advantages:
- Span large stretches: A key advantage of long-read sequencing is its ability to span the largest repeat stretches, sequencing them from start to finish in a single read. This eliminates the need for computational assembly, simplifying downstream analysis.
- Complex alleles: Long-read sequencing can adeptly handle complex extended alleles, especially when repeated sequences are interrupted multiple times—a situation where short-read sequencing and some diagnostic methods may fail.
- Direct Detection: Since no PCR amplification is required, potential bias is eliminated, especially in GC-rich regions. Furthermore, this allows direct detection of base modifications, as demonstrated by nanopore technology, enabling comprehensive repeat extension interrogation.
Limitations:
- Error rates: Current long-read sequencing technologies tend to have higher error rates than short-read platforms. But as technology continues to improve, this gap is expected to narrow.
While short-read sequencing remains a trusted method for many genomic analyzes due to its high accuracy and throughput, its limitations become apparent when dealing with large repeat expansions. In contrast, long-read sequencing, with its ability to span large amplifications and detect complex alleles, emerges as a promising tool for comprehensive repeat expansion analysis.
Straglr: A New Software Tool for Repeat Expansion
More than 40 diseases have been found to be caused by STR amplification. Repeat expansion may also be an underlying mechanism in other rare diseases that are currently unexplained. Efficiently searching extensions anywhere in the genome would greatly facilitate the identification of novel disease-associated STR loci, but genome-wide searches of such extensions using existing long-read sequencing genotyping software require interrogation of hundreds of thousands of bands. Annotated sites. Straglr was developed to enhance the advantages of long-read sequencing. As researchers and clinicians strive to understand and combat diseases caused by STR expansion, tools like Straglr play an integral role in that effort.
Straglr operates on a two-pronged approach. Initially, it identifies insertions consisting of TRs. After this identification, it genotypes the extension of the markers. This approach not only saves computational resources but also paves the way for the discovery of previously unannotated loci. This is a shift from traditional approaches where genotyping non-expanded TR loci is time- and resource-intensive.
The efficiency of this software has been proven through rigorous testing. Simulated and real long-read data demonstrate the strength of Straglr for STR-extended genotyping. Its promise is not limited to reaffirming known data; it also has the potential to uncover new disease-associated STR loci. Given the importance of studying STR extensions—including cryptic extensions known and not yet associated with disease.
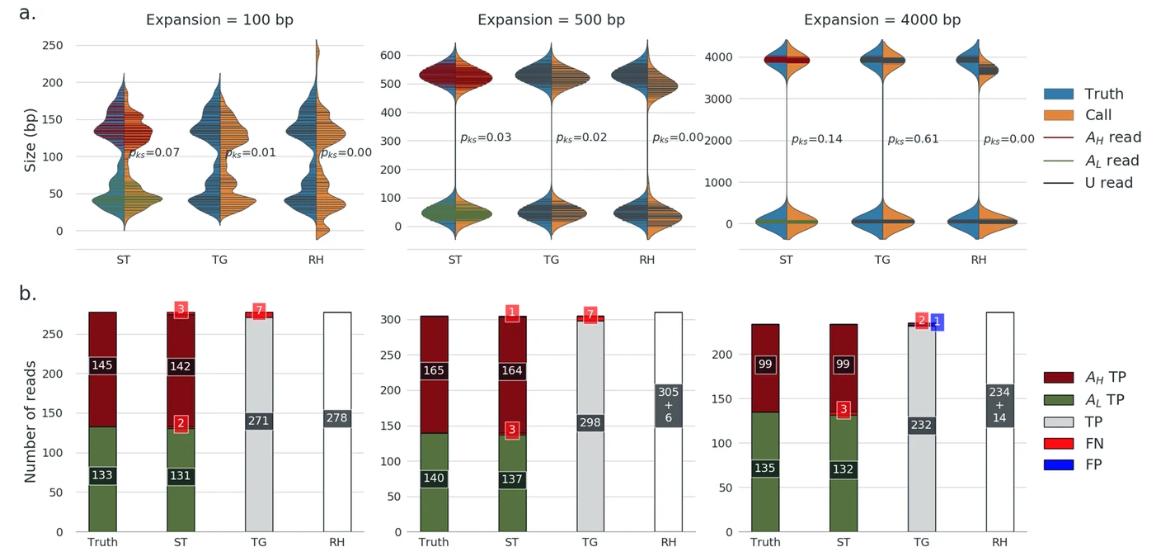 Genotyping benchmark (simulated data): repeat capture. (Chiu et al., 2021)
Genotyping benchmark (simulated data): repeat capture. (Chiu et al., 2021)
References
- Paulson, Henry. "Repeat expansion diseases." Handbook of clinical neurology 147 (2018): 105-123.
- Bahlo, Melanie, et al. "Recent advances in the detection of repeat expansions with short-read next-generation sequencing." F1000Research 7 (2018).
- Chiu, Readman, et al. "Straglr: discovering and genotyping tandem repeat expansions using whole genome long-read sequences." Genome Biology 22.1 (2021): 224.
Related Services
PacBio SMRT Sequencing
Oxford Nanopore Sequencing
Human Long-Amplicon Sequencing
Animal/Plant Long Amplicon Sequencing
Genome-Wide Association Study (GWAS)
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment