We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Services

Transcriptomics with Long-Read Sequencing

circRNA Full-Length Identification and Quantification Service

Full-Length Transcript Sequencing (Iso-Seq)

Nanopore Direct RNA Sequencing
Nanopore Full-Length cDNA Sequencing

Full-Length Transcriptome Profiling Combined with Long- and Short-Read Sequencing

Full-Length Spatial-Temporal Transcriptome Profiling

Single-Cell Full-Length Transcriptome Sequencing
Application of Long-Read Sequencing Technology in Single-Cell Transcriptomics
At a glance:
- Technological Advancements
- Current Applications and Innovations
- Key Applications in Biomedical Research
- Case Study
- Summary
In an era characterized by rapid technological advancements, research in the field of omics is experiencing unprecedented growth. Single-cell transcriptomics has emerged as a pivotal tool in life sciences, enabling a comprehensive understanding of gene expression at the individual cell level. This technology elucidates the differences among various cell types and captures dynamic changes during development and disease processes. The advent of long-read (LR) transcriptome sequencing technology has expanded the scope of single-cell transcriptomics beyond mere gene-level quantification, allowing for the detailed analysis of cell-specific transcription start sites and differential transcript isoforms. This technological evolution facilitates a deeper understanding of the complexity of the transcriptome at the isoform level.
Technological Advancements
Designated as the "Method of the Year" in 2022, long-read sequencing has markedly transformed contemporary life sciences and biomedical research. This technique offers researchers unprecedented opportunities to delve into transcriptomic complexity, presenting a comprehensive view of the transcriptome by capturing full-length isoforms. Its significance stems from its capacity to address the limitations inherent in short-read sequencing, which frequently fails to accurately reconstruct full-length transcripts due to its reliance on fragmented reads.
The convergence of third-generation full-length transcriptome sequencing technology with single-cell transcriptomics represents an emerging and promising research frontier. This integrative approach not only facilitates the identification of novel isoforms but also enhances our understanding of transcriptional regulation at a more detailed level. Long-read sequencing enables the detection of transcript variants that might otherwise remain elusive, thus advancing our comprehension of cellular heterogeneity and uncovering potential therapeutic targets.
Current Applications and Innovations
CD Genomics has recently introduced a novel sequencing platform that combines Iso-Seq and MAS-Seq technologies. This platform significantly enhances throughput, achieving a 16-fold increase compared to traditional methods. The application of this high-throughput, long-read sequencing technology enables the detailed characterization of transcriptomes at the single-cell level. It allows for the accurate mapping of full-length transcripts, providing a more complete and accurate representation of the transcriptomic landscape.
This technological innovation holds promise for advancing our understanding of cellular processes and disease mechanisms. By capturing the full spectrum of transcript isoforms, researchers can investigate the functional consequences of alternative splicing events, which play crucial roles in various biological processes and diseases, including cancer. The ability to analyze transcript isoforms in a high-throughput manner opens new avenues for the discovery of biomarkers and the development of targeted therapies.
Service you may intersted in
Key Applications in Biomedical Research
The integration of third-generation full-length sequencing technology with single-cell transcriptomics has catalyzed substantial advancements in the study of diverse biomedical phenomena. This synergistic approach proves particularly effective in identifying gene fusions and cell-specific transcriptional isoforms, which are pivotal in driving diseases such as cancer. Additionally, it elucidates the mechanisms underlying alternative splicing, a process that generates a variety of functionally distinct proteins from a single gene.
Oncology and Disease Mechanisms:
The detection of gene fusions and alternative isoforms is critically important in oncology research, as these genetic features often serve as biomarkers for tumorigenesis. This technology has been instrumental in elucidating the molecular mechanisms underlying various cancers, including colorectal, breast, liver, and ovarian cancers. Through comprehensive characterization of transcript variants, researchers are afforded new perspectives on the intricate regulatory networks that drive tumor development and progression.
Neurodevelopment and Immunology:
In the realm of neurodevelopment, full-length single-cell transcriptomics has been instrumental in mapping the transcriptomic landscape of the developing human brain. This includes detailed analyses of cell type-specific splicing events that may contribute to neurological disorders. Similarly, in immunology, the technology enables the comprehensive profiling of immune cell populations, facilitating the identification of unique isoforms associated with immune responses and disease states.
Case Study
Case Study 1: Developmental Isoform Diversity in the Human Neocortex
Title: Developmental Isoform Diversity in the Human Neocortex Informs Neuropsychiatric Risk Mechanisms
Abstract
The intricate molecular and genetic regulation of human brain development, marked by widespread RNA splicing, holds significant implications for neuropsychiatric disorders. Despite the ubiquity of splicing events, the specific contributions of cell type-specific splicing and isoform diversity to brain development remain poorly understood. This study leverages single-cell isoform sequencing (scIso-Seq) to conduct a comprehensive analysis of full-length transcriptomes in the germinal zone (GZ) and cortical plate (CP) regions of the developing human neocortex during gestation. The generated data culminate in the establishment of an unprecedented cell type-specific isoform expression atlas, providing novel insights into the mechanisms underlying brain development and associated disease processes.
Introduction
Human brain development is orchestrated by complex molecular and genetic processes, prominently involving RNA splicing. Splicing plays a pivotal role in the diversification of the transcriptome, particularly in the context of neuronal differentiation and function. Neuropsychiatric disorders have been associated with aberrations in these splicing mechanisms, suggesting that a comprehensive understanding of isoform diversity is crucial for elucidating disease etiology.
Methods
The study utilized scIso-Seq to capture and analyze the full-length transcriptomes from both tissue and single-cell levels in the GZ and CP regions of the developing human neocortex. This technique enables the identification of full-length isoforms, providing a more complete picture of transcriptomic diversity compared to traditional short-read sequencing methods.
Results
A total of 214,516 unique transcript isoforms were identified, of which 72.6% were novel. This extensive diversity underscores the complexity of transcriptomic regulation during neocortical development. The data revealed that RNA-binding proteins play a significant role in regulating these isoforms, which in turn define the cellular characteristics of the developing neocortex. Furthermore, through gene annotation of these isoforms, thousands of candidate transcripts associated with rare de novo risk variants for neuropsychiatric disorders were discovered, shedding light on potential genetic mechanisms underlying these conditions.
Discussion
The findings highlight the importance of isoform diversity in brain development and its potential link to neuropsychiatric diseases. The identification of novel isoforms and their regulation by RNA-binding proteins suggests a complex layer of post-transcriptional control that contributes to cellular differentiation and function in the neocortex. Moreover, the association of specific isoforms with rare genetic variants implicates these transcripts in the etiology of neuropsychiatric disorders, providing new avenues for research and therapeutic intervention.
Conclusion
This study demonstrates the power of scIso-Seq in elucidating the full-length transcriptomic landscape of the developing human neocortex. The comprehensive identification of novel isoforms and their regulatory mechanisms provides valuable insights into brain development and the genetic basis of neuropsychiatric disorders. Future research leveraging these findings may lead to improved diagnostic and therapeutic strategies for these complex conditions.
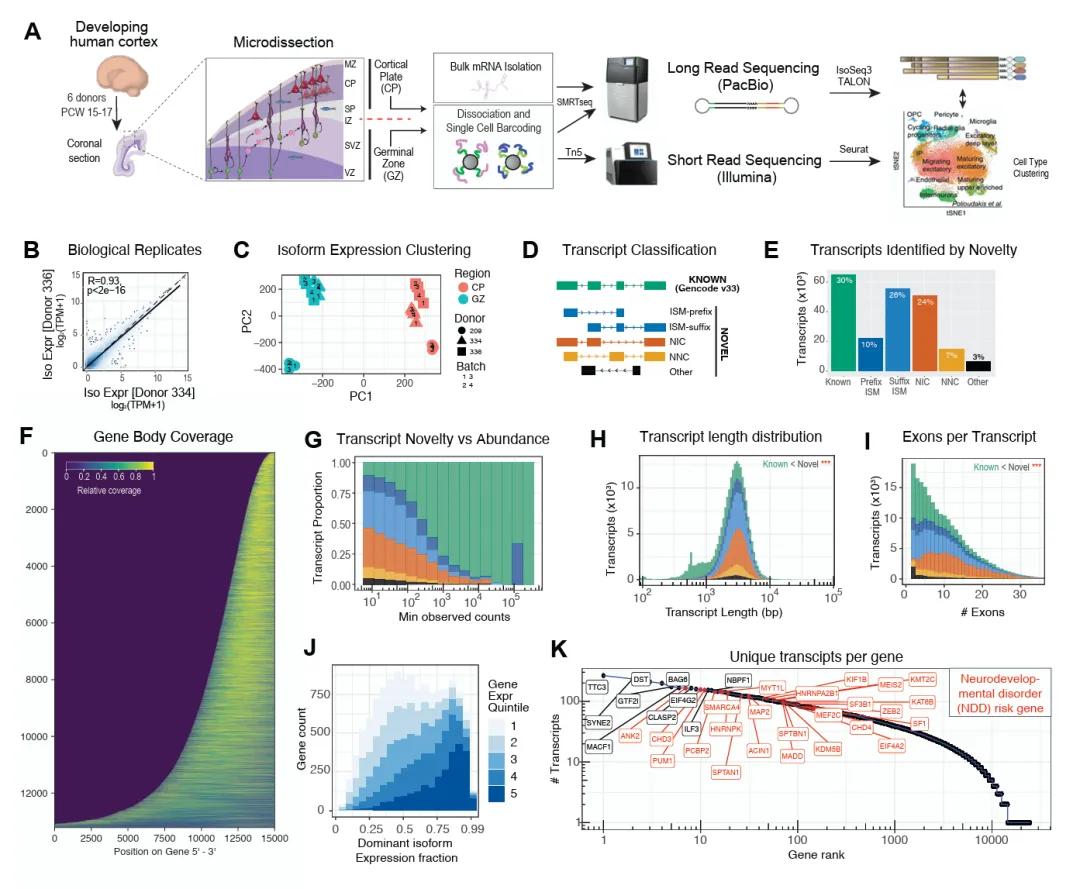 Figure 1: Cell type-specific full-length transcriptome of the human neocortex during mid-gestation.
Figure 1: Cell type-specific full-length transcriptome of the human neocortex during mid-gestation.
Case Study 2: Full-Length Transcriptome Sequencing Reveals Splicing Isoform Complexity and Diversity in Breast Cancer
Title: A Comprehensive Long-Read Isoform Analysis Platform and Sequencing Resource for Breast Cancer
Abstract
This study leverages Iso-Seq to conduct full-length transcriptome sequencing on the breast cancer transcriptome, previously considered extensively studied. The cohort includes four normal human breast samples and twenty-six tumor samples, comprising two cell lines and two normal breast tissues, as well as thirteen primary breast tumor biopsy tissues (three hormone receptor-positive ER+/PR+, three HER2+, and seven triple-negative breast cancers), nine patient-derived xenograft (PDX) tumors, and four breast cancer cell lines. Utilizing third-generation full-length transcriptome sequencing, thousands of previously unannotated isoforms were identified, approximately 30% of which affect protein-coding exons. These isoforms are predicted to alter protein localization and function, resulting in the identification of 3059 breast tumor-specific splicing events, with thirty-five events significantly associated with patient survival. Through comprehensive full-length single-cell transcriptome analysis, the study elucidates the complexity of previously unknown splicing events, their subtype specificity, and clinical relevance in breast cancer, providing a rich resource for immuno-oncology therapeutic targets. The authors conclude that Iso-Seq is particularly suitable for discovering isoforms containing novel tumor-immune targets, including those encoding cell surface proteins, which can be targeted by specific monoclonal antibodies for therapeutic use or as scaffolds for chimeric antigen receptor (CAR) T cells. Additionally, isoforms generate peptides potentially useful in vaccination strategies, possibly in combination with immune checkpoint inhibitors.
Introduction
Breast cancer, a heterogeneous disease, exhibits significant transcriptomic diversity, contributing to its varied clinical presentations and therapeutic responses. RNA splicing plays a crucial role in generating transcriptomic diversity, influencing both gene expression and protein function. Despite extensive research, many aspects of the breast cancer transcriptome remain unexplored, particularly regarding the diversity and functional implications of splicing isoforms. This study aims to uncover the complexity of splicing isoforms in breast cancer, focusing on identifying clinically relevant events that could inform new therapeutic strategies.
Methods
The study employed Iso-Seq, a third-generation sequencing technology, to capture full-length transcriptomes from a cohort comprising normal and cancerous breast tissues. The cohort included four normal samples (two cell lines and two normal breast tissues) and twenty-six tumor samples, encompassing a range of subtypes: ER+/PR+, HER2+, and triple-negative breast cancers. The inclusion of PDX tumors and breast cancer cell lines enabled a comprehensive analysis of splicing events across different breast cancer models.
Results
The analysis identified thousands of previously unannotated isoforms, approximately 30% of which involve protein-coding exons. These isoforms are predicted to affect protein function and localization, highlighting the functional diversity generated by splicing in breast cancer. Notably, 3059 breast tumor-specific splicing events were identified, with thirty-five events significantly associated with patient survival, underscoring their potential clinical relevance. The study also revealed the complexity of splicing events in breast cancer, with distinct patterns observed across different subtypes. This subtype specificity suggests that splicing isoforms may serve as biomarkers for cancer classification and prognosis.
Discussion
The identification of novel splicing isoforms in breast cancer provides insights into the molecular mechanisms underlying tumor heterogeneity. The discovery of isoforms with potential immune-related functions is particularly noteworthy, as these could serve as targets for immunotherapy. For example, isoforms encoding cell surface proteins may be targeted by specific monoclonal antibodies, which can be developed into therapeutic agents or CAR T cell therapies. Furthermore, the study highlights the potential of isoform-specific peptides as components of vaccination strategies, particularly in conjunction with immune checkpoint inhibitors.
Conclusion
This comprehensive analysis of the breast cancer transcriptome, utilizing full-length sequencing, reveals the extensive complexity and clinical relevance of splicing isoforms. The findings provide a valuable resource for the development of new therapeutic strategies, particularly in the field of immuno-oncology. Iso-Seq emerges as a powerful tool for the discovery of novel isoforms, offering new opportunities for targeted therapy development and personalized medicine in breast cancer.
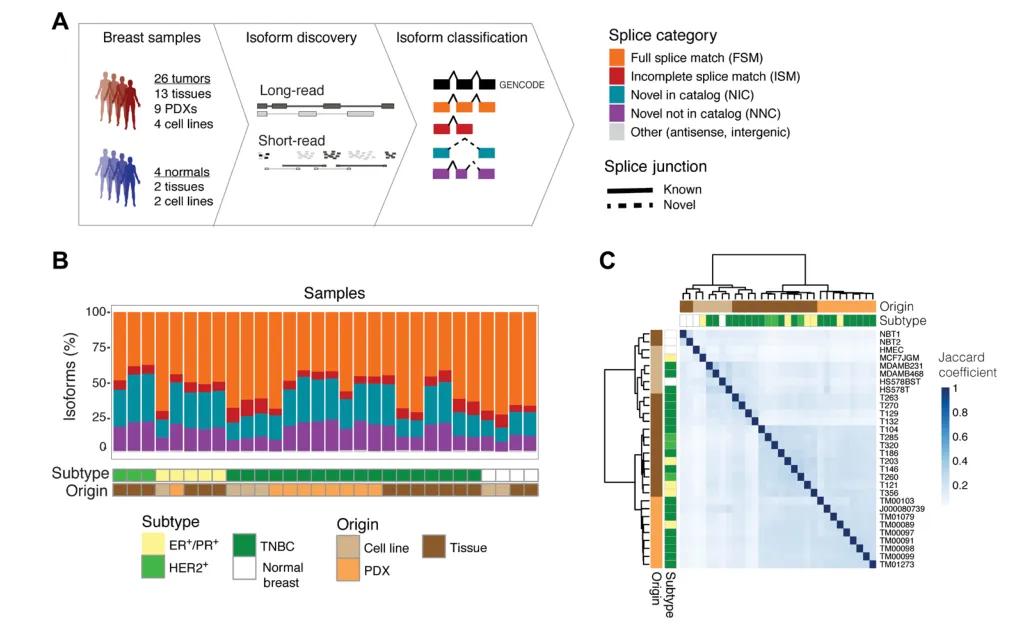 Figure 2: Full-length transcriptome study of breast cancer.
Figure 2: Full-length transcriptome study of breast cancer.
Case Study 3: Identification of Transcript Isoforms and Genomic Alterations in Ovarian Cancer through Full-Length Single-Cell Transcriptome Sequencing
Title: Detection of Isoforms and Genomic Alterations by High-Throughput Full-Length Single-Cell RNA Sequencing in Ovarian Cancer
Abstract
This study employs full-length single-cell transcriptome sequencing to analyze clinical samples from three patients with high-grade serous ovarian carcinoma (HGSOC), including five samples (two adjacent non-cancerous samples). PacBio sequencing technology was utilized, achieving a depth of 12,000 reads per cell. The analysis identified 152,000 isoforms, with over 52,000 previously unreported, demonstrating the efficacy of long-read sequencing in detecting rare or low-abundance transcripts. Additionally, the study identified cell type-specific isoforms and polyadenylation sites in tumor and mesothelial cells. It was observed that during tumor metastasis, a subset of mesothelial cells undergoes transformation into cancer-associated fibroblasts via the TGF-β/miR-29/collagen axis. The study also uncovered multiple gene fusion events, including a validated IGF2BP2::TESPA1 fusion, and further investigated the associated genomic alterations. The findings underscore the growing importance of third-generation full-length single-cell transcriptome sequencing in oncology and personalized medicine.
Introduction
High-grade serous ovarian carcinoma (HGSOC) is a highly aggressive and heterogeneous malignancy. Traditional transcriptomic analyses have been limited in their ability to fully capture the complexity of transcript isoforms and genomic alterations. This study aims to utilize advanced single-cell RNA sequencing technologies to provide a comprehensive understanding of the transcriptomic landscape in HGSOC, focusing on the identification of rare isoforms and gene fusion events.
Methods
Clinical samples were obtained from three HGSOC patients, including five samples, two of which were adjacent non-cancerous tissues. Full-length single-cell RNA sequencing was performed using PacBio technology, achieving an unprecedented sequencing depth of 12,000 reads per cell. The study utilized bioinformatics pipelines to identify and characterize transcript isoforms and gene fusion events, as well as to detect cell type-specific expression patterns and polyadenylation sites.
Results
A total of 152,000 isoforms were identified, with more than 52,000 being novel. This highlights the capability of long-read sequencing to uncover previously uncharacterized transcript variants, particularly those present at low abundance. Specific isoforms and polyadenylation sites were delineated in tumor and mesothelial cells, revealing distinct expression patterns. Notably, a subset of mesothelial cells was found to transdifferentiate into cancer-associated fibroblasts through the TGF-β/miR-29/collagen axis, a process implicated in tumor metastasis.
The study also detected several gene fusion events, including the IGF2BP2::TESPA1 fusion, which was experimentally validated. Further genomic analyses revealed associated chromosomal rearrangements, underscoring the utility of single-cell RNA sequencing in identifying clinically relevant genomic alterations.
Discussion
The application of full-length single-cell transcriptome sequencing in this study has provided critical insights into the transcriptomic diversity and genomic landscape of HGSOC. The identification of novel isoforms and gene fusions emphasizes the complexity of the disease and the need for advanced sequencing technologies to fully elucidate these features. The discovery of mesothelial cell transdifferentiation into cancer-associated fibroblasts highlights a potential mechanism of tumor progression and metastasis, offering new avenues for therapeutic intervention.
Conclusion
The study demonstrates the power of third-generation full-length single-cell RNA sequencing in oncology research, providing a detailed view of transcript isoforms and genomic alterations in ovarian cancer. The findings suggest that this technology will become increasingly valuable in the field of personalized medicine, offering potential for the identification of novel therapeutic targets and biomarkers.
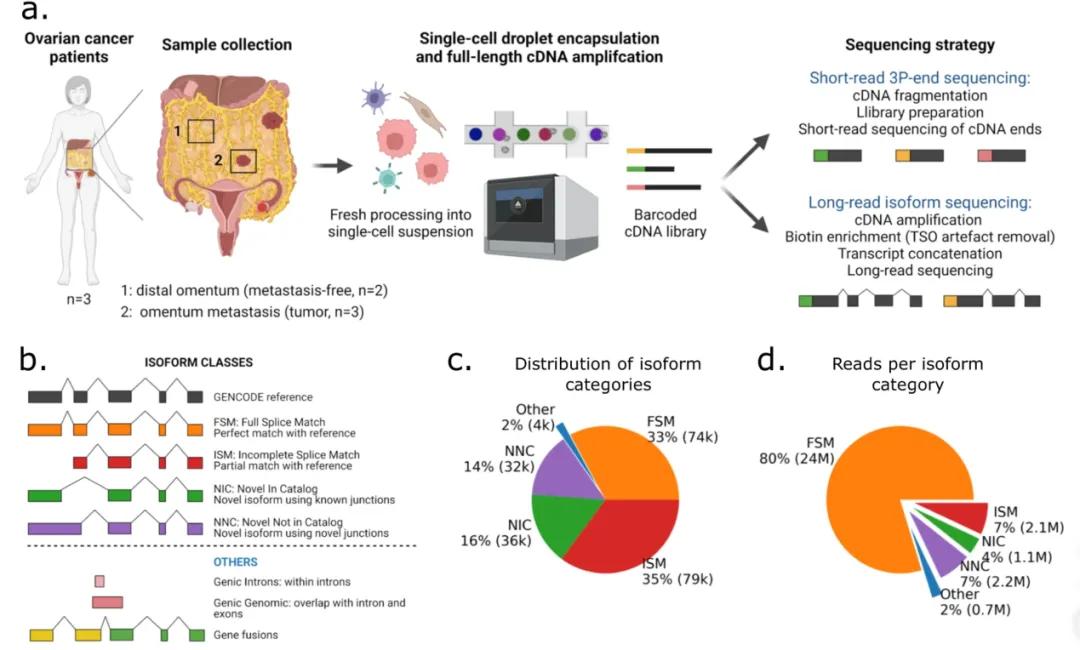 Figure 3: Full-length single-cell transcriptome analysis of ovarian cancer.
Figure 3: Full-length single-cell transcriptome analysis of ovarian cancer.
Case Study 4: Long-Read Single-Cell Sequencing Facilitates Research on Reproductive Disorders
Title: Long-Read Single-Cell Sequencing Reveals the Transcriptional Landscape of Spermatogenesis in Obstructive Azoospermia and Sertoli Cell-Only Patients
Abstract
This study utilizes long-read single-cell transcriptome sequencing to delineate the transcriptional landscape associated with spermatogenesis in patients diagnosed with obstructive azoospermia (OA) and non-obstructive azoospermia (NOA), specifically Sertoli cell-only syndrome. Single cells were isolated from testicular biopsies obtained from OA (n = 3) and NOA (n = 3) patients. Comparative analysis was performed using both long-read sequencing (PacBio) and short-read sequencing technologies (OA n = 6, NOA n = 6). In the OA cohort, 130,426 long-read transcripts were detected, including 100,517 novel transcripts and 29,909 known transcripts. Similarly, in the NOA cohort, 49,508 long-read transcripts were identified, comprising 26,002 novel and 23,506 known transcripts. Additionally, the study identified 36,373 and 1,642 novel genes in OA and NOA patients, respectively. Notably, specific expression of long-read transcripts in germ and somatic cells during normal spermatogenesis was observed. These findings provide valuable insights into the molecular underpinnings of human spermatogenesis and potential therapeutic avenues for male infertility.
Introduction
Azoospermia, defined by the complete absence of spermatozoa in the ejaculate, represents a prevalent cause of male infertility. It is categorized into OA and NOA, the latter encompassing conditions such as Sertoli cell-only syndrome. Traditional transcriptomic techniques have been limited in their ability to capture the full spectrum of transcript isoforms, particularly those pertinent to spermatogenesis. This study utilizes long-read single-cell RNA sequencing to delineate the transcriptional landscape of spermatogenesis, offering a comprehensive analysis of both novel and known transcripts in patients with OA and NOA.
Methods
Testicular biopsies were obtained from patients diagnosed with OA and NOA. Single cells were isolated and subjected to long-read single-cell RNA sequencing using PacBio technology. For comparative purposes, short-read sequencing was also performed. Data analysis focused on identifying novel and known transcripts, as well as novel gene discovery. The study employed rigorous bioinformatics pipelines to ensure accurate transcript annotation and classification.
Results
In the OA group, long-read single-cell sequencing identified 130,426 transcripts, including 100,517 novel and 29,909 known transcripts. In the NOA group, 49,508 transcripts were identified, consisting of 26,002 novel and 23,506 known transcripts. The study further revealed the presence of 36,373 novel genes in OA patients and 1,642 novel genes in NOA patients. The long-read sequencing technology enabled the detection of cell type-specific transcripts, providing a detailed view of the transcriptional landscape during normal and impaired spermatogenesis.
The analysis uncovered specific expression patterns of long-read transcripts in germ cells and somatic cells, highlighting the potential functional significance of these transcripts in the regulation of spermatogenesis. Additionally, the study identified several key transcripts that may serve as biomarkers for diagnosing and differentiating between OA and NOA.
Discussion
The comprehensive transcriptomic profiling achieved in this study underscores the utility of long-read single-cell RNA sequencing in uncovering the complexities of spermatogenesis. The identification of a vast array of novel transcripts and genes not only expands our understanding of the genetic basis of azoospermia but also opens new avenues for therapeutic intervention. The distinct expression patterns observed in germ and somatic cells provide insights into the molecular mechanisms governing normal and disrupted spermatogenesis, offering potential targets for future research and clinical applications.
Conclusion
This study underscores the efficacy of long-read single-cell RNA sequencing in offering a detailed and nuanced understanding of the transcriptional landscape underlying spermatogenesis in patients with OA and NOA. The identification of novel transcripts and genes makes a significant contribution to the field of reproductive biology and male infertility, revealing potential biomarkers and therapeutic targets. Future research should prioritize the functional validation of these findings and the development of targeted therapies to address male infertility.
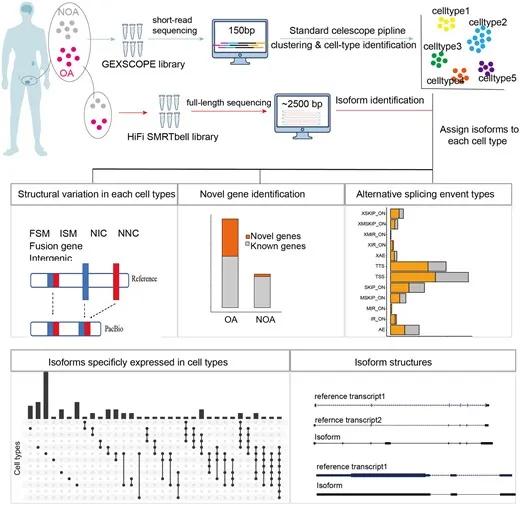 Figure 4: Full-length transcriptome study of obstructive azoospermia.
Figure 4: Full-length transcriptome study of obstructive azoospermia.
Summary
The integration of single-cell transcriptomics with long-read sequencing technologies enables the exploration of more complex biomedical questions and unveils more specific events and mechanisms.
This article explores the applications of long-read sequencing in single-cell transcriptomics across various domains of biomedical research. Long-read sequencing has revolutionized transcriptomic studies by overcoming the limitations of short-read sequencing, offering a comprehensive view of transcript isoforms and enabling detailed analysis of cell-specific transcriptional landscapes.
In the realm of oncology, long-read sequencing has proven invaluable in identifying novel splicing isoforms and gene fusions, providing insights into tumor heterogeneity and potential therapeutic targets. Notably, studies on breast cancer and ovarian cancer have revealed previously unannotated isoforms and genomic alterations, highlighting the technology's role in advancing personalized medicine and immuno-oncology.
The application of long-read single-cell sequencing in reproductive disorders, specifically in obstructive and non-obstructive azoospermia, has unveiled a wealth of novel and known transcripts, enhancing our understanding of spermatogenesis. This approach offers new opportunities for diagnosing and treating male infertility by identifying key biomarkers and transcriptional regulators.
Overall, long-read single-cell transcriptome sequencing represents a powerful tool for elucidating the complexities of gene expression, with far-reaching implications for cancer research, neurodevelopment, and reproductive biology. This synergistic approach represents a significant advancement in the field of life sciences, offering new breakthroughs and propelling research to new heights.
References
- Conte M I, Fuentes-Trillo A, Conde C D. Opportunities and tradeoffs in single-cell transcriptomic technologies. Trends in Genetics, 2023.
- Patowary A, Zhang P, Jops C, et al. Developmental isoform diversity in the human neocortex informs neuropsychiatric risk mechanisms. bioRxiv, 2023: 2023.03. 25.534016.
- Veiga D F T, Nesta A, Zhao Y, et al. A comprehensive long-read isoform analysis platform and sequencing resource for breast cancer. Science Advances, 2022, 8(3): eabg6711.
- Dondi A, Lischetti U, Jacob F, et al. Detection of isoforms and genomic alterations by high-throughput full-length single-cell RNA sequencing in ovarian cancer. Nature Communications, 2023, 14(1): 7780.
- Wu X, Lu M, Yun D, et al. Long-read single-cell sequencing reveals the transcriptional landscape of spermatogenesis in obstructive azoospermia and Sertoli cell-only patients. QJM: An International Journal of Medicine, 2024: hcae009.
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment