

We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Nanopore Sequencing 101: Bioinformatics Analysis of ONT Data
At a glance:
- Data Analysis Pipeline for ONT Sequencing
- Base Calling and Base Modification Detection
- Detecting DNA and RNA Modifications in ONT Sequencing
- Error Correction and Polishing
- De Novo Assembly and Genome Polishing
- Alignment and Variant Calling
Oxford Nanopore Technologies (ONT) sequencing has witnessed significant progress in recent years, becoming a key player in the genomics field. As the technology matures, so does the bioinformatics analysis of ONT data. Researchers have been diligently developing specialized tools and algorithms to better utilize the unique characteristics of ONT data, such as long read lengths and ionic current signals. This article explores the latest bioinformatics advancements that enable enhanced base calling, base modification detection, error correction, assembly, and alignment of ONT data.
Data Analysis Pipeline for ONT Sequencing
The bioinformatics analysis of ONT data typically involves a multi-step pipeline to transform raw electrical signals into meaningful genomic information. The pipeline includes base calling, error correction, alignment, variant calling, and additional steps for specialized analyses, such as detecting modifications and assessing transcriptome complexity.
 Workflow of bioinformatics analysis – CD Genomics
Workflow of bioinformatics analysis – CD Genomics
Explore our Oxford Nanopore Sequencing Data Analysis Service for more information.
Base Calling and Base Modification Detection
Base calling is a fundamental step in ONT data analysis, converting the raw ionic current signals into DNA base sequences. Early versions of base callers had relatively high error rates, hindering downstream analyses. However, with continuous improvements, modern base callers, such as Guppy and Chiron, have significantly enhanced accuracy and now offer real-time base calling capabilities.
Furthermore, ONT technology is uniquely suited to detect epigenetic modifications, such as DNA methylation. Specialized algorithms, including Tombo and DeepSignal, have been developed to identify base modifications by analyzing specific changes in the ionic current signal associated with modified bases. This epigenetic information is crucial for understanding gene regulation and other biological processes.
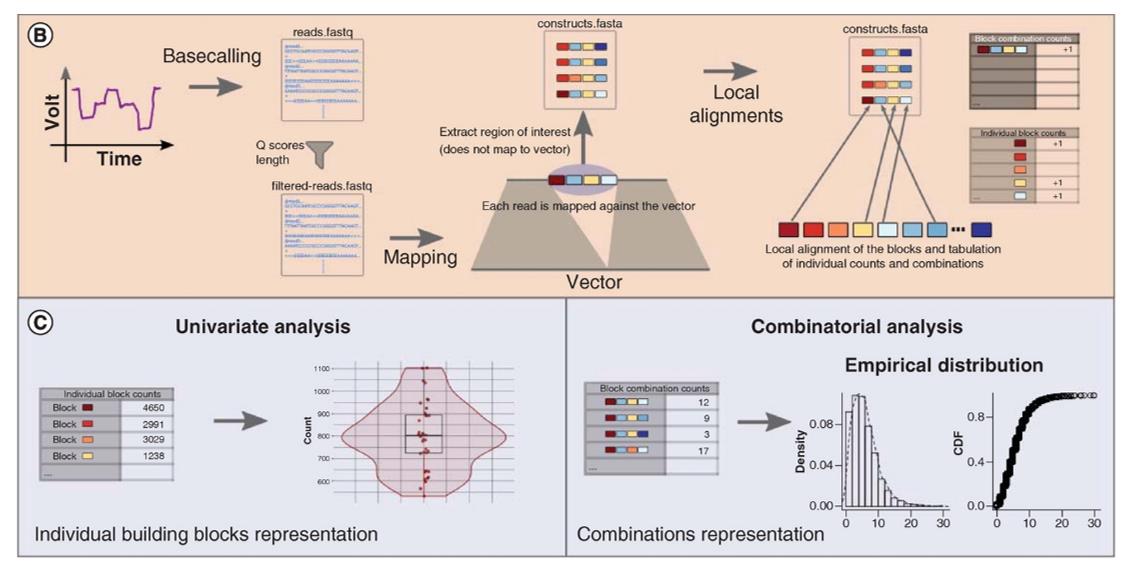 Bioinformatics pipeline of ONT sequencing. (Lood et al., 2020)
Bioinformatics pipeline of ONT sequencing. (Lood et al., 2020)
Detecting DNA and RNA Modifications in ONT Sequencing
One of the key advantages of Oxford Nanopore Technologies (ONT) sequencing is its ability to directly detect DNA and RNA modifications. By distinguishing the unique current shifts caused by modified bases from those of unmodified bases, ONT sequencing offers insights into epigenetic modifications and post-transcriptional RNA modifications. In this section, we explore the methodologies and tools developed for the detection of DNA and RNA modifications using ONT sequencing data.
CD Genomics offers Epigenetics and Methylation Analysis Using Long-Read Sequencing for both DNA and RNA modifications.
DNA Modification Detection
ONT sequencing enables the direct detection of certain DNA modifications, such as 5-methylcytosine (5mC), 6-methyladenine (6mA), and N4-methylcytosine (4mC), at different levels of resolution, ranging from bulk-level detection to the single-molecule level. Several tools have been developed to identify DNA modifications from ONT data:
- Nanoraw: Integrated into the Tombo software package, Nanoraw was the first tool to detect DNA modifications, including 5mC, 6mA, and 4mC, from ONT data.
- Nanopolish: This tool specializes in detecting 5mC modifications in DNA sequences and has been widely used for accurate modification calling.
- SignalAlign: SignalAlign detects 5mC, 5-hydroxymethylcytosine (5hmC), and 6mA modifications in DNA sequences, providing valuable information on the epigenetic landscape.
- mCaller, DeepMod, and DeepSignal: These tools have been developed to identify both 5mC and 6mA modifications, contributing to a comprehensive view of DNA modifications in ONT sequencing data.
- NanoMod: NanoMod is another tool designed to detect 5mC and 6mA modifications in DNA, further enhancing the accuracy and coverage of modification detection.
RNA Modification Detection
Detecting RNA modifications directly using ONT sequencing has also shown promise, although the resolution varies, and single-nucleotide resolution at the single-molecule level is yet to be demonstrated. In the past, PacBio sequencing was used to detect N6-methyladenosine (m6A) modifications in RNA molecules. More recently, ONT direct RNA sequencing has generated robust data of reasonable quality, paving the way for the detection of RNA modifications. Several pilot studies have successfully detected bulk-level RNA modifications using various methodologies:
- EpiNano and ELIGOS: These tools examine error distribution profiles or current signals to detect m6A and 5-methoxyuridine (5moU) modifications in RNA.
- Tombo extension and MINES: By analyzing current signals, these tools have been used to identify m6A modifications in RNA molecules.
Although these pilot studies have detected bulk-level RNA modifications, achieving single-nucleotide resolution at the single-molecule level remains a challenge.
Error Correction and Polishing
While the average accuracy of Oxford Nanopore Technologies (ONT) sequencing is improving, certain subsets of reads or read fragments still exhibit very low accuracy. The error rates of both 1D reads and 2D/1D2 reads remain higher than those of short reads generated by next-generation sequencing technologies. As a result, error correction is a critical step applied before many downstream analyses, such as genome assembly and gene isoform identification. Error correction helps rescue reads for higher sensitivity and improves the quality of the results, including breakpoint determination at single-nucleotide resolution.
There are two main types of error correction algorithms used in ONT sequencing data analysis:
- Self-Correction: Self-correction algorithms employ graph-based approaches to produce consensus sequences among different molecules from the same origins. For instance, tools like Canu and LoRMA perform self-correction to improve the accuracy of 1D reads and 2D/1D2 reads generated from the same molecules.
- Hybrid Correction: Hybrid correction algorithms utilize high-accuracy short reads to correct long reads. These algorithms can be alignment-based, graph-based, or a combination of both. Examples of hybrid correction tools include LSC, Nanocorr, LorDEC, and HALC. By leveraging the information from short reads, hybrid correction methods significantly reduce the error rate of long reads.
Recently, benchmark studies have demonstrated the efficacy of existing hybrid error correction tools, such as FMLRC, LSC, and LorDEC, along with sufficient short-read coverage, to reduce the long-read error rate to a level (approximately 1-4%) similar to that of short reads. On the other hand, self-correction reduces the error rate to approximately 3-6%, which may be attributed to non-random systematic errors in ONT data.
De Novo Assembly and Genome Polishing
The long read lengths offered by ONT sequencing make it ideal for de novo genome assembly of complex organisms. De novo assembly tools like Canu and Flye have been adapted to specifically handle ONT data. These tools take advantage of the long reads to span repetitive regions and resolve complex genomic structures, providing more contiguous and accurate genome assemblies.
Genome polishing with ONT data, using tools like Pilon, further refines the assembled genome, correcting errors and misassemblies, leading to high-quality reference genomes.
Alignment and Variant Calling
Aligning error-prone long reads, such as those generated by Oxford Nanopore Technologies (ONT) sequencing, poses unique challenges due to the high error rates and increased read lengths compared to traditional short-read data. In response to the rise of long-read sequencing technologies, specialized aligners have been developed to effectively handle the distinct characteristics of error-prone long reads.
- GraphMap: The First ONT-Specific Aligner
In 2016, GraphMap became the first aligner explicitly designed for ONT reads. It was initially motivated by PacBio data but also demonstrated effectiveness on ONT data. GraphMap employs a progressive refinement approach to handle high error rates and utilizes fast graph traversal algorithms for high-speed and precise alignment of long reads.
- Minimap2: Efficient and Accurate Aligner for ONT Reads
Minimap2 was developed to cater to the increasing read lengths beyond 100 kb in ONT sequencing. Using a seed-chain-align procedure, minimap2 achieved remarkable performance, running faster than other long-read aligners like LAST, NGMLR, and GraphMap, while still maintaining high accuracy. Notably, minimap2 also supports splice-aware alignment for ONT cDNA or direct RNA-sequencing reads, making it well-suited for transcriptomic applications.
- Splice-Aware Alignment for Transcriptome Data
In the realm of transcriptome data, aligners like GMAP (published in 2005) and the short-read aligner STAR, adapted for long reads, have been widely used for splice-aware alignment of error-prone transcriptome long reads to reference genomes. These aligners help identify exon-exon junctions and detect alternative splicing events, providing valuable insights into gene expression and isoform diversity.
- Other Aligners for ONT Transcriptome Data
Additional aligners have been developed specifically for ONT transcriptome data, including Graphmap2 and deSALT. Graphmap2 has demonstrated superior alignment rates over minimap2, particularly for ONT direct RNA-sequencing reads with dense base modifications, making it well-suited for analyzing heavily modified RNA molecules.
Reference
- Lood, Cédric, et al. "Quality control and statistical evaluation of combinatorial DNA libraries using nanopore sequencing." BioTechniques 69.5 (2020): 379-383.
Related Services
Oxford Nanopore Sequencing Data Analysis
Epigenetics and Methylation Analysis Using Long-Read Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment