Long-Read vs Short-Read Sequencing
At a glance:
- Long-Read Sequencing
- Short-Read Sequencing
- Long Read vs. Short Read Sequencing: A Detailed Comparison
- Hybrid Use of Long-Read and Short-Read Sequencing
- When to Choose Long-Read Sequencing
- When to Choose Short-Read Sequencing
- Understanding the Advantages of Long-Read Sequencing
- Challenges and Limitations of Long-Read Sequencing
- Conclusion
Sequencing platforms can be divided into long-read and short-read types, each essential for genomic research. This article examines the operational principles of both platforms and highlights their differences, offering researchers valuable guidance in choosing the most appropriate sequencing technology for their specific research objectives.
1. Long-Read Sequencing
Long-read sequencing is a cutting-edge tool in modern DNA analysis, offering significant advancements over traditional methods by addressing complex genomic challenges with greater detail. Unlike short-read techniques, long-read sequencing can analyze DNA fragments ranging from thousands to hundreds of thousands of base pairs, excelling in areas where previous methods struggle, such as resolving repetitive sequences, detecting complex structural variations, and uncovering previously ambiguous genomic regions. This technology is not just an improvement—it's a revolutionary tool that significantly enhances our understanding of genetics.
1.1 Sequencing Principle
The core advantage of long-read sequencing lies in its ability to read long fragments of DNA directly without the need for extensive fragmentation or amplification, making it ideal for sequencing complex and repetitive regions. There are two primary techniques used for long-read sequencing:
Single-Molecule Real-Time (SMRT) Sequencing: Developed by Pacific Biosciences (PacBio), SMRT sequencing captures real-time nucleotide incorporation events as a DNA polymerase synthesizes a complementary strand. A zero-mode waveguide (ZMW) helps detect nucleotide incorporation events with high precision, allowing the technology to generate accurate long reads without prior amplification.
Nanopore Sequencing: Pioneered by Oxford Nanopore Technologies, this method passes single DNA molecules through nanopores embedded in a membrane. The ion current across the nanopore changes as nucleotides pass through, and these current fluctuations are analyzed to determine the DNA sequence. This approach is unique in its ability to sequence extremely long DNA strands, with read lengths extending into the millions of base pairs.
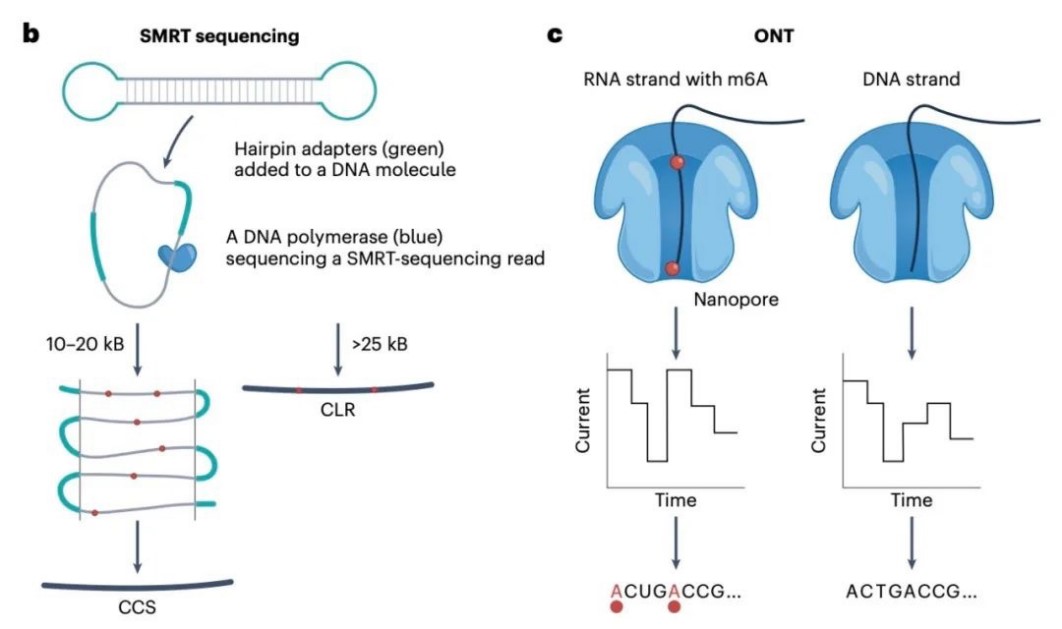 Figure 1.Long-read sequencing principles of SMRT and ONT.(Foord,et.al,2023)
Figure 1.Long-read sequencing principles of SMRT and ONT.(Foord,et.al,2023)
1.2 Technology Platforms
Key platforms for long-read sequencing include:
PacBio HiFi Sequencing: Known for its high-precision HiFi sequencing, it is suitable for gene mutation analysis with high accuracy requirements.
Oxford Nanopore Technologies: Support for extremely long read lengths (up to millions of bases), and portable devices such as MinION enable on-site sequencing.
Bionano Genomics: While primarily used for structural variation detection, Bionano Genomics complements long-read sequencing by mapping ultra-long DNA molecules and aiding in genome assembly validation.
| Technology Platform | Maximum Read Length | Accuracy (Quality Score) | Main Advantages | Representative Equipment |
| PacBio | 100 kb+ | High (HiFi: 99.9%) | High-fidelity long reads, suitable for complex genomes | Sequel IIe |
| ONT | 1 Mb+ | Moderate (85%-95%) | Extremely long reads, highly portable | MinION |
| Bionano | >150 kb | Not applicable (no sequence information) | Ultra-long fragments for structural variation and optical validation | Saphyr |
Learn More
Overview of Long-read Sequencing
2. Short-Read Sequencing
Short-read sequencing is the modern genetic research, turning small pieces of DNA into valuable scientific discoveries. By analyzing DNA fragments between 50 and 300 base pairs with great accuracy, this method has transformed the way we study genes. Its biggest strength is its ability to handle large amounts of data efficiently and affordably, making it a reliable choice for clinical studies, population genetics, and detailed molecular research. When faced with complicated challenges, short-read sequencing proves to be a flexible and powerful tool, uncovering important stories hidden in our DNA.
2.1 Sequencing Principle
Short-read sequencing is a multi-step process designed to analyze DNA fragments efficiently and accurately. The workflow typically includes the following key steps:
Step1 Fragmentation: Genomic DNA is fragmented into smaller segments, generally through mechanical shearing or enzymatic digestion, to produce DNA fragments of appropriate lengths for sequencing.
Step2 Adapter Ligation: Synthetic adapters are ligated to the ends of the fragmented DNA. These adapters enable the fragments to bind to sequencing platforms and serve as priming sites for amplification and sequencing.
Step3 Amplification and Sequencing: The prepared DNA fragments undergo amplification to generate clusters of identical sequences. Platforms such as Illumina utilize bridge amplification, while others like Roche 454 employ emulsion PCR. Once amplified, sequencing is performed using techniques such as reversible dye terminators (Illumina) or pyrosequencing (Roche 454), enabling high-throughput analysis of the DNA fragments.
2.2 Technology Platforms
Several key platforms dominate the short-read sequencing market:
Illumina NovaSeq 6000: This platform provides massive throughput, making it suitable for large-scale sequencing projects, including whole-genome sequencing, exome sequencing, and transcriptome analysis. NovaSeq supports read lengths ranging from 50 bp to 300 bp, with high accuracy and depth of coverage.
Thermo Fisher Ion Torrent: Using semiconductor technology, Ion Torrent sequences DNA by detecting changes in pH as nucleotides are incorporated. It offers a fast and affordable sequencing option, particularly useful for targeted sequencing and amplicon-based applications.
MGI Tech DNBSEQ: An alternative to Illumina's platforms, DNBSEQ offers competitive throughput and reduced sequencing costs, making it suitable for large-scale studies.
3. Long Read vs. Short Read Sequencing: A Detailed Comparison
Long-read and short-read sequencing have inherent differences, making them suited for distinct applications. Below is a comparative analysis of these two sequencing methods:
| Aspect | Long-Read Sequencing | Short-Read Sequencing |
| Read Length | Thousands to hundreds of thousands of base pairs | 50–300 base pairs |
| Accuracy | High (PacBio HiFi); Moderate (Nanopore) | Very high (error rates < 0.1%) |
| Cost | Higher per base | Lower per base |
| Throughput | Moderate | High |
| Assembly | Ideal for de novo assembly | Challenging for complex genomes |
| Structural Variations | Superior resolution of structural variants | Limited sensitivity for structural variants |
| Scalability | Suitable for complex and long-term projects | Excellent for high-throughput projects |
| Portability | Portable options (e.g., MinION) | Requires dedicated lab infrastructure |
Case Study:
Mark Sheehan and colleagues conducted an in-depth study comparing the performance of short-read sequencing and long-read sequencing in evaluating the integration of adeno-associated virus (AAV) vectors. AAV, a critical vector in gene therapy, primarily exists as an episomal element in the cell nucleus. However, approximately 0.1%–0.5% of AAV vectors may integrate into the host genome, posing potential risks such as genomic instability or oncogenesis. To assess these integration events, the research team employed Targeted Enrichment Sequencing (TES) to analyze integration patterns. They utilized short-read sequencing on the Illumina platform and long-read sequencing on the PacBio platform, investigating samples from AAV-treated monkey liver tissues, lentivirus-treated cell lines, stable cell lines, and artificial controls. Their findings demonstrated that short-read sequencing, with its high coverage, identified more integration sites and excelled in quantitatively analyzing hotspots and clonal expansion events. In contrast, long-read sequencing revealed vector rearrangements in 4%–40% of integration sites and directly measured integration lengths and complex recombination patterns, proving advantageous for resolving intricate integration events. Overall, short-read sequencing is better suited for high-precision quantitative analysis, while long-read sequencing is more effective for studying complex genomic structures and rearrangements. Selecting the appropriate sequencing technology based on specific research needs is essential for comprehensively evaluating AAV vector integration characteristics.
 Figure 2.Comparison of long-read and short-read TES methods for analyzing AAV integration into the host genome.(Sheehan et.al ,2024)
Figure 2.Comparison of long-read and short-read TES methods for analyzing AAV integration into the host genome.(Sheehan et.al ,2024)
4. Hybrid Use of Long-Read and Short-Read Sequencing
This collaboration goes beyond just combining technologies; it forms a comprehensive scientific strategy. By integrating both sequencing methods, researchers can transform fragmented genetic data into a cohesive and detailed narrative. This approach uncovers the intricate architectural features of biological complexity, offering new insights into the genetic makeup of life. Rather than seeing each sequencing technique as a standalone tool, this synergy provides a more complete understanding, where limitations are overcome, and deeper scientific discoveries are made.
Case Study:
Gounot et.al utilized a hybrid assembly approach that combined short-read sequencing, long-read sequencing, and Hi-C technology to analyze the gut microbiomes of 109 individuals from three distinct ethnic groups in Singapore. The integration of these sequencing methods allowed for the generation of 4,497 metagenome-assembled genomes (MAGs), a significant increase compared to the 2,789 MAGs produced using only short reads. This approach revealed the presence of 70 previously uncharacterized microbial species, more than 3,400 novel strains, and thousands of biosynthetic gene clusters (BGCs) that were not present in existing databases. The research also uncovered a distinct and non-characteristic microbial diversity in the Southeast Asian population, highlighting the importance of population-specific microbiome data for disease research and drug discovery. By merging short and long reads, the study achieved improved assembly continuity, with the hybrid assembly showing a significantly higher N50 value and greater genome completeness. The findings emphasize the value of hybrid sequencing approaches for enhancing microbial diversity reference libraries and their potential in precision medicine and biosynthetic exploration, particularly in underrepresented populations such as those in Southeast Asia.
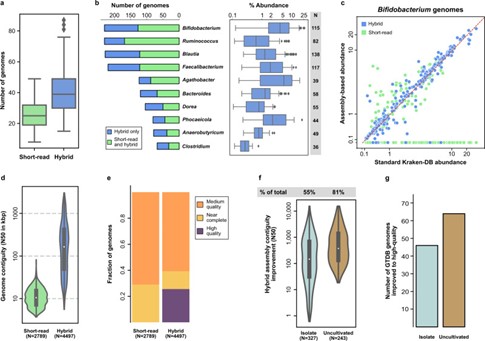 Figure 3.Assembly strategy for high-quality microbiome references.(Gounot et.al ,2022)
Figure 3.Assembly strategy for high-quality microbiome references.(Gounot et.al ,2022)
5. When to Choose Long-Read Sequencing
Indeed, long-read sequencing technologies have significantly advanced the field of genomics by addressing many of the limitations of short-read sequencing methods. Here's a bit more detail on each of the key applications you've mentioned:
De Novo Genome Assembly: When assembling genomes de novo, long-read sequencing technology proves indispensable, particularly for non-model organisms or genomes lacking detailed annotation. Consider the challenges faced by scientists tackling a complex genome riddled with extensive repetitive regions and large gaps. This is precisely where the advantages of long-read sequencing come to the forefront. For instance, researchers successfully utilized long-read sequencing to assemble the genome of the coffee plant, a genome previously deemed difficult to resolve due to its highly repetitive DNA sequences(Cheng,2017). The ability of long-read sequencing to span these vast gaps and repetitive regions enables the construction of contiguous and accurate genomic structures. This capability not only uncovers the genetic diversity of the coffee plant but also provides valuable insights for crop improvement and stress resistance research.
Analysis of Complex Genome Structure:Long-read sequencing excels in addressing intricate genomic structures, particularly in detecting repetitive elements, telomeric and centromeric regions, and substantial structural variations, including large insertions and deletions (indels). For instance, by leveraging long-read sequencing, scientists conducted an in-depth examination of the maize (Zea mays) genome, uncovering large gene insertion events that had previously eluded detection(Li, Z,2022). These complex genomic features often remain unresolved or inaccurately characterized when analyzed with short-read sequencing techniques.
Haplotype Phasing: The use of long-read sequencing facilitates accurate haplotype phasing, improving the ability to resolve allele-specific inheritance patterns. This feature is especially crucial in population genetics research, where precise haplotypic data is indispensable.A key application is the use of single-sperm long-read sequencing for haplotype identification, which has been enhanced over the years to offer greater accuracy in characterizing genetic variants, including structural variations (50bp-10kb). This approach provides a more efficient and precise method for haplotyping compared to traditional methods reliant on short reads or parent-based sequencing(Tang et.al,2023).
Full-Length Transcript Sequencing: Full-length transcript sequencing using long-read technologies enables the analysis of entire RNA transcripts, offering detailed information on transcript structure, splicing patterns, isoform variability, and alternative splicing processes. This depth of information is essential for uncovering the regulatory mechanisms of gene expression and advancing functional genomic studies.
Learn more
Application of Long Read Sequencing: Genome Assembly
6. When to Choose Short-Read Sequencing
Short-read sequencing remains the preferred choice for many applications due to its cost efficiency and high scalability. Specific use cases include:
- High-Throughput Studies: Short-read sequencing is optimal for large-scale projects, such as population genetics studies, clinical genomic analyses, and other high-throughput applications where processing thousands of samples is required.
- Variant Detection: This technology provides exceptional accuracy for identifying single-nucleotide variants (SNVs) and small insertions or deletions (indels) in well-characterized genomic regions.
- RNA Sequencing for Gene Expression Analysis: Short-read sequencing offers precise quantification of gene expression levels and transcript-level insights, making it ideal for deep transcriptome analysis in functional genomics.
7. Understanding the Advantages of Long-Read Sequencing
Long-read sequencing offers distinct advantages that are vital for addressing the challenges of complex genomic research:
- De Novo Genome Assembly: The generation of extensive, high-fidelity reads makes long-read sequencing indispensable for constructing genomes de novo, particularly for non-model organisms lacking established reference genomes.
- Detection of Structural Variants and Indels: This approach effectively identifies intricate structural variants and large insertions or deletions that are often overlooked by short-read methods.
- Resolution of Repetitive and Complex Genomic Regions: Long-read sequencing uniquely enables the analysis of highly repetitive regions, including centromeres, telomeres, and extensive segmental duplications, which remain inaccessible to short-read sequencing technologies.
8. Challenges and Limitations of Long-Read Sequencing
Despite its advantages, long-read sequencing faces certain challenges:
Higher Cost: Long-read sequencing technologies like PacBio and Oxford Nanopore are more expensive per base compared to short-read platforms, making them less suitable for large-scale, high-throughput studies.
Higher Error Rates (Nanopore): While PacBio HiFi sequencing boasts high accuracy, Oxford Nanopore's error rates are higher, although recent algorithmic improvements have significantly reduced this issue.
Throughput: While PacBio and Nanopore have high scalability, they do not yet match the throughput of Illumina or Ion Torrent platforms, particularly in high-throughput projects requiring thousands of samples to be processed quickly.
9. Conclusion
Long-read and short-read sequencing have revolutionized genomic studies, each offering distinct advantages suited to specific research objectives. Short-read sequencing is widely utilized for its cost-effectiveness, scalability, and exceptional accuracy in detecting genetic variants. Conversely, long-read technologies, including PacBio HiFi and Oxford Nanopore, excel at accurately identifying large structural variants and elucidating complex genomic rearrangements often linked to conditions like cancer and neurological disorders. By enabling comprehensive mapping of structural variations across entire genomes, these methods provide critical insights into the genetic underpinnings of such diseases.
CD Genomics offers state-of-the-art sequencing solutions that integrate the strengths of both long-read and short-read platforms. Whether addressing structural variant resolution, performing de novo genome assembly, or investigating gene expression patterns, our expert team is dedicated to delivering reliable, high-quality results tailored to your research objectives. Reach out to CD Genomics to discover how our advanced sequencing technologies can empower your scientific advancements.
References
- Foord, C., Hsu, J., Jarroux, J., Hu, W., Belchikov, N., Pollard, S., He, Y., Joglekar, A., & Tilgner, H. U. (2023). The variables on RNA molecules: concert or cacophony? Answers in long-read sequencing. Nature methods, 20(1), 20–24. https://doi.org/10.1038/s41592-022-01715-9
- Sheehan, M., Kumpf, S. W., Qian, J., Rubitski, D. M., Oziolor, E., & Lanz, T. A. (2024). Comparison and cross-validation of long-read and short-read target-enrichment sequencing methods to assess AAV vector integration into host genome. Molecular therapy. Methods & clinical development, 32(4), 101352. https://doi.org/10.1016/j.omtm.2024.101352
- Gounot, J. S., Chia, M., Bertrand, D., Saw, W. Y., Ravikrishnan, A., Low, A., Ding, Y., Ng, A. H. Q., Tan, L. W. L., Teo, Y. Y., Seedorf, H., & Nagarajan, N. (2022). Genome-centric analysis of short and long read metagenomes reveals uncharacterized microbiome diversity in Southeast Asians. Nature communications, 13(1), 6044. https://doi.org/10.1038/s41467-022-33782-z
- Cheng, B., Furtado, A., & Henry, R. J. (2017). Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. GigaScience, 6(11), 1–13. https://doi.org/10.1093/gigascience/gix086
- Li, Z., Han, L., Luo, Z., & Li, L. (2022). Single-molecule long-read sequencing reveals extensive genomic and transcriptomic variation between maize and its wild relative teosinte (Zea mays ssp. parviglumis). Molecular ecology resources, 22(1), 272–282. https://doi.org/10.1111/1755-0998.13454
- Xie, H., Li, W., Guo, Y., Su, X., Chen, K., Wen, L., & Tang, F. (2023). Long-read-based single sperm genome sequencing for chromosome-wide haplotype phasing of both SNPs and SVs. Nucleic acids research, 51(15), 8020–8034. https://doi.org/10.1093/nar/gkad532
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment