

We are dedicated to providing outstanding customer service and being reachable at all times.


What is a Consensus Sequence?
At a glance:
- Overview
- What is a Consensus Sequence
- Consensus Sequences: Functional Significance in Bioinformatics
- Realistic Examples of Consensus Sequences
- Visualizing Consensus Sequences
- Consensus Sequences VS. Sequence Patterns
Overview
Organisms have similar and different characteristics. The genome or genetic composition of an organism is important when studying the relationships between organisms and categorizing them. Gene sequences can change at any time. They undergo mutations, deletions, translocations, and inversions that gradually change the sequence over time. Despite these forces, some sequences remain unchanged, such as conserved and consensus sequences.
The generation of shared sequences is a fundamental task in many bioinformatics analyses. Our Oxford Nanopore Technologies (ONT) system for nanopore sequencing enables high-throughput, long-read sequencing of DNA and RNA samples to analyze nucleotide or amino acid sequences that are consensus between homologous regions of different but related DNA or RNA or protein sequences. This sequence-based bioinformatics analysis can provide us with useful information about the evolutionary history of an organism, the approximate age of sequence families (e.g., transposon families), the unique variation at each nucleotide/amino acid position, the identification of splice sites (pre-mRNAs), etc.
What is a Consensus Sequence
A consensus sequence is a common nucleotide sequence or amino acid sequence found in highly conserved regions of DNA or RNA or proteins. It represents the result of multiple sequence comparisons in which related sequences are compared with each other and similar sequence motifs are calculated.
 Extracting a consensus sequence from a multiple sequence alignment. (Sternke M et al., 2020)
Extracting a consensus sequence from a multiple sequence alignment. (Sternke M et al., 2020)
Consensus Sequences: Functional Significance in Bioinformatics
Bioinformatics utilizes the concept of shared sequences as a cornerstone for a large number of analyses. The potential applications are very diverse and cover different areas of biological research.
Evolution
A major use of consensus sequences lies in the field of phylogenetic studies. These sequences can provide insight into the evolutionary history of a group of species. For example, comparing the shared sequences of different species can reveal genetic differences that indicate how each of these species evolved over-time.
Prediction of gene function
The presence of certain consensus sequences can often predict gene function. A prime example is transcription factor binding motifs within promoter regions. The presence of shared sequences in a particular motif suggests that specific transcription factors may bind there, thus indicating the likely function of the gene.
Identification of Molecular Mechanisms
Consensus sequences can help reveal complex molecular mechanisms, such as selective splicing. They can help obtain accurate mRNA sequences from expressed sequence tags (ESTs), discover differences between paralogous homologous sequences, and even eliminate spurious single nucleotide polymorphisms (SNPs) associated with alignment and/or sequencing errors.
Phenotypic Variation
When studying populations, consensus sequences can help identify single base substitutions that may affect phenotype. As such, they are a valuable tool for understanding the type and distribution of alleles in a population.
Realistic Examples of Consensus Sequences
To better understand the relevance of consensus sequences, one needs to consider the TATA box - a consensus promoter sequence known to initiate gene transcription in archaea and eukaryotes. This TATA box sequence (TATAWAW), where the "W" stands for adenine (A) or thymine (T), provides a clear example of how consensus sequences can have real-world significance.
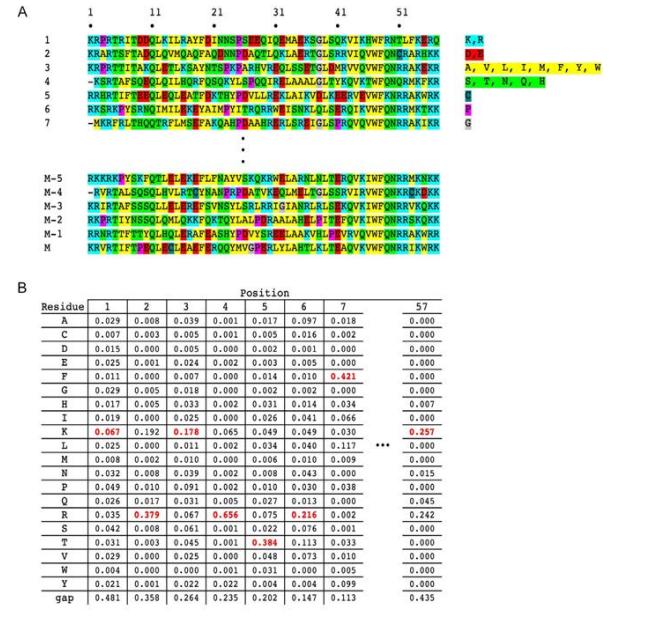
Visualizing Consensus Sequences
A well-established tool for visualizing consensus sequences is the sequence identity. These graphical representations illustrate the consistency and diversity of sequences. In sequence logos, the size of the letters is proportional to the frequency of a particular nucleotide or amino acid at a particular position, providing a direct way to understand sequence information.
Consensus Sequences VS. Sequence Patterns
Despite their similarities, consensus sequences and sequence patterns are distinct entities. Consensus sequences represent the linear order of nucleotides or amino acids for a given sequence family, but sequence patterns are evolutionarily conserved sequences that can represent hidden functional properties of a sequence. For example, subtle base differences that may alter the topology of the original sequence binding site can only be detected by conserved patterns.
References
- Sternke M, Tripp K W, Barrick D. The use of consensus sequence information to engineer stability and activity in proteins[M]//Methods in enzymology. Academic Press, 2020, 643: 149-179.
- Vierstraete, Andy R., and Bart P. Braeckman. "Amplicon_sorter: A tool for reference‐free amplicon sorting based on sequence similarity and for building consensus sequences." Ecology and Evolution 12.3 (2022): e8603.
Related Services
Oxford Nanopore Sequencing
Oxford Nanopore Pre-Made Library Sequencing
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment