We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Applications of Iso-Seq in the Medical Field
At a glance:
- Explore Region-Specific Expression of Transcripts
- Uncovering the Regulatory Mechanisms of Growth and Development
- Identifying Specific Isoforms in Diseases/Tumors
- Conclusion
Isoform Sequencing (Iso-Seq) is a powerful technology developed by Pacific Biosciences that leverages long-read sequencing to generate full-length cDNA sequences. This method eliminates the need for transcriptome reconstruction, providing direct access to complete transcript isoforms, including alternative splicing events, transcription start sites, and polyadenylation sites. Here are some key applications of Iso-Seq in the medical field.
Explore Region-Specific Expression of Transcripts
Full-length transcriptome sequencing can provide a comprehensive view of transcripts, including their sequence, structure, expression, etc. By selecting samples from different tissues or different regions of the same tissue, it is possible to observe the transcript structural characteristics and the changes in gene/transcript expression with the region, thereby understanding the transcript structure and expression characteristics of different cell types or tissue regions.
Iso -Seq identifies region-specific transcript isoforms in the human brain
This study used long-read RNA sequencing technology to analyze region-specific gene isoforms in three different brain regions (cerebellum, hypothalamus, and temporal cortex) in the human brain. Although isoforms that may be artifacts were excluded after rigorous screening, it was found that more than half of the isoforms were not registered in the GENCODE reference database in multiple samples. The study paid special attention to genes that expressed different major isoforms in each brain region, even if the overall expression levels of these genes were similar, and found that many such genes (such as GAS7) may have different roles in dendritic spine and neuron formation in each region. In addition, the study also found that DNA methylation may drive the differences in isoform expression in different regions to some extent. These findings emphasize the importance of analyzing isoforms expressed in disease-related sites.
Background
Different brain regions are affected in neurological and neuropsychiatric diseases, and differences in gene expression patterns may explain this mechanism. Previous studies were mostly based on short-read sequences or microarray data, and their isoforms were inferred only from short-read sequences. Advances in long-read sequencing technology have made it possible to read the entire transcript from 5' to 3' untranslated regions (UTRs) and polyA tails at one time, thereby capturing transcript features more accurately.
Methods
Sample source: The study used hindbrain samples from four male deceased persons aged over 50 years old with no brain lesions or infections, and extracted RNA from the cerebellum, hypothalamus, and temporal cortex.
Sequencing technology: Iso-Seq sequencing was performed using the Sequel IIe system from PacBio.
Data processing: Cluster analysis was performed using IsoSeq v3 software, and quality control and filtering were performed using SQANTI3 to exclude possible artifacts such as nonsense-mediated decay and false cDNA molecules.
Methylation analysis: Combined with DNA methylation data, the MeDeCom method was used to estimate cellular composition and analyze the impact of DNA methylation on alternative splicing.
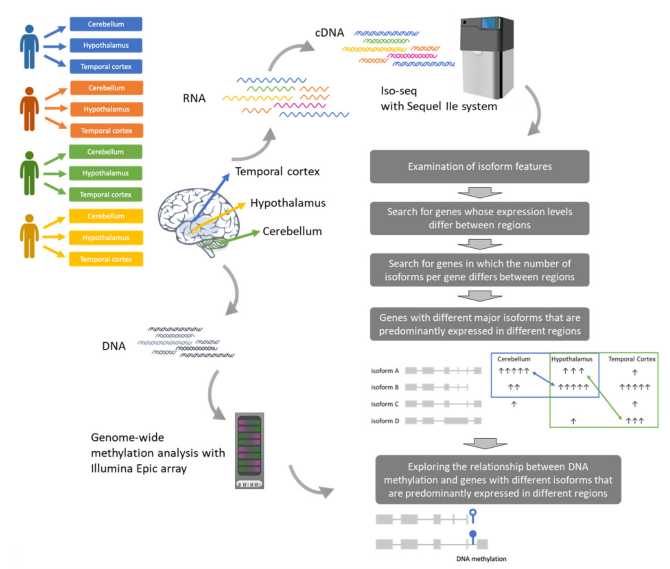 Figure 1.Graphical abstract and analytical flow of Iso-seq data.(Shimada, M et al. 2024)
Figure 1.Graphical abstract and analytical flow of Iso-seq data.(Shimada, M et al. 2024)
Results
Iso-Seq studies of three brain regions found that an average of 27,643 isoforms were identified per sample, and a total of 66,860 non-redundant isoforms were identified. These isoforms were annotated to 14,382 known genes and 52 unknown genes. Comparing the number and structural characteristics of isoforms in different brain regions, it was found that the number of isoforms in the cerebellum was significantly lower than that in the hypothalamus and temporal cortex, while the number of exons of a single gene in the cerebellum was higher than that in other brain regions. This study identified the regional specific isoforms in different brain regions. The main subtypes of gene isoforms in different brain regions were explored, and the ratio (%) of the expression level of a single isoform to the total gene expression level was calculated by Iso-Seq sequencing, and the ratios and rankings of different brain regions were compared, and isoforms with a ratio difference of more than 20% (P < 0.05) were searched. The results identified 193 isoforms of 145 genes in the cerebellum and hypothalamus, 147 isoforms of 114 genes in the cerebellum and temporal cortex, and 55 isoforms of 47 genes in the hypothalamus and temporal cortex. The study found that there are differences in the expression of isoforms in different brain regions, and these different isoforms that are mainly expressed in specific brain regions play key roles in biological processes.
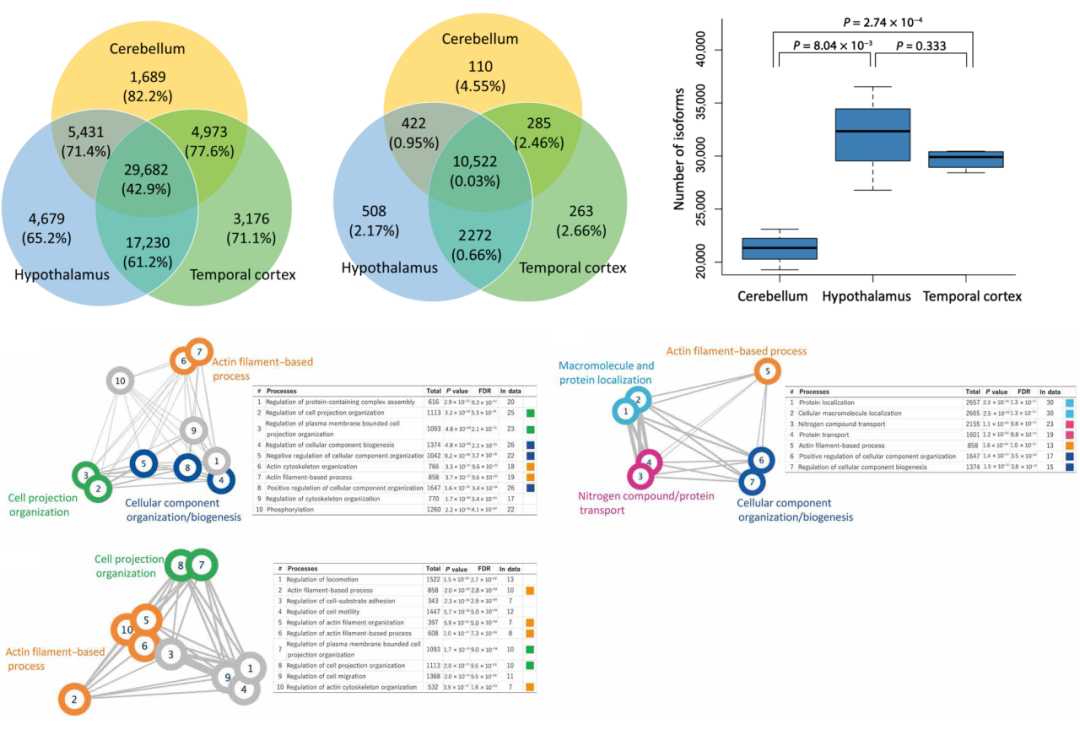 Figure 2.Isoforms structure features of brain regions and gene function enrichment of major subtypes in brain regions.(Shimada, M et al. 2024)
Figure 2.Isoforms structure features of brain regions and gene function enrichment of major subtypes in brain regions.(Shimada, M et al. 2024)
Uncovering the Regulatory Mechanisms of Growth and Development
By selecting samples at different developmental time points, we can observe the changes in transcript structural characteristics and gene/transcript expression over time, thereby revealing the dynamic changes in cell differentiation, development or disease progression, and thus understanding the dynamic regulatory mechanism of the gene expression regulatory network.
Iso-seq reveals mRNA remodeling mechanisms in human oocyte-to-embryo transformation
This article studies the poly(A) tail-mediated post-transcriptional regulation of maternal mRNA during human oocyte-to-embryo transition (OET). During OET, due to the lack of transcription, post-transcriptional regulation of maternal mRNA is essential for early embryonic development. The dynamic changes of poly(A) tails play an important role in OET in many species, but it is still unclear in humans.
Methods
The poly(A)-inclusive RNA isoform sequencing (PAIso-seq) technique was used to study the dynamic changes of poly(A) tails in human oocytes and early embryos. The role of genes such as BTG4, TUT4, TUT7, TENT4A, and TENT4B in the regulation of poly(A) tails was studied by siRNA-mediated gene knockdown experiments. The results of PAIso-seq were verified using PAIso-seq2 technology, and the dynamic changes of poly(A) tails were further analyzed.
Results
The length of the poly(A) tail and internal non-A residues during the transition from oocyte to embryo are highly dynamic. During oocyte maturation, the relative abundance of transcripts with poly(A) tails of 25-60 nt decreased in metaphase I and further decreased in metaphase II, indicating that global deadenylation of maternal mRNA occurred. After fertilization, the relative abundance of transcripts with 12-60 nt poly(A) tails increased in one-cell, two-cell, and four-cell embryos, indicating global polyadenylation. The overall trend is that the polyA tail becomes shorter from germinal vesicle to metaphase II, and there is a trend of re-polyA after fertilization, and polyA becomes longer. The study reveals that poly(A) tail-mediated maternal mRNA remodeling is essential for human embryonic development.
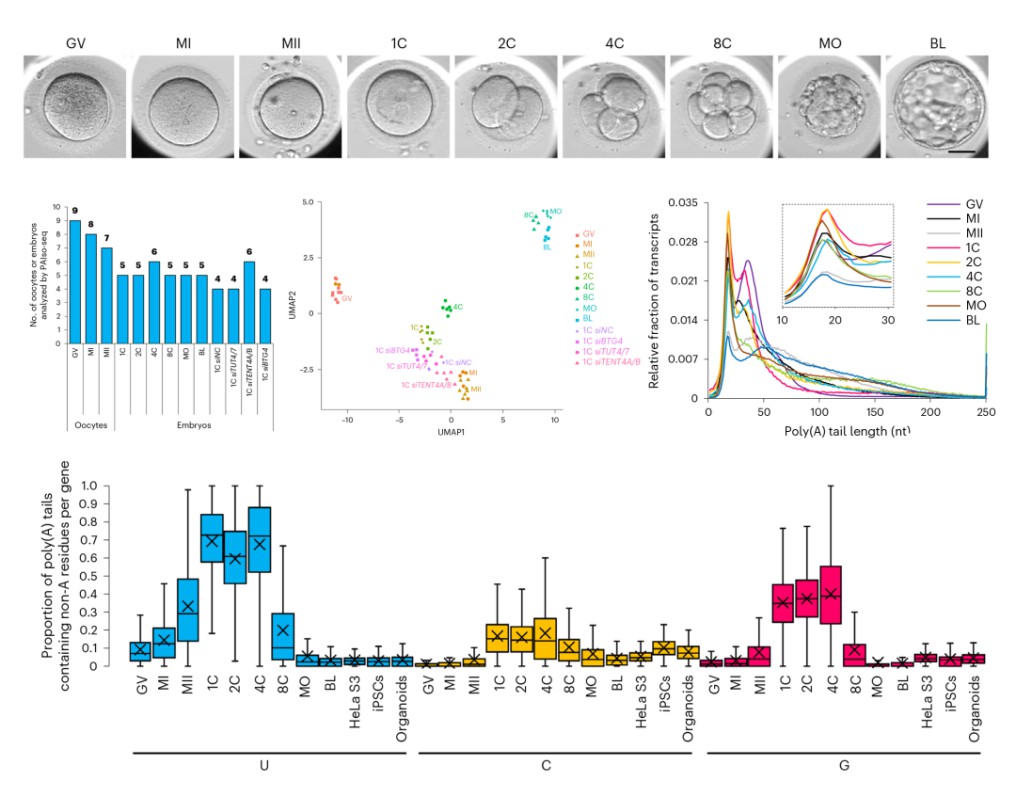 Figure 3.Sample selection and different periods of poly (A) length and poly (A) tail in the non-A base homosexual love situation.(Liu, Y et al. 2023)
Figure 3.Sample selection and different periods of poly (A) length and poly (A) tail in the non-A base homosexual love situation.(Liu, Y et al. 2023)
Identifying Specific Isoforms in Diseases/Tumors
Obtain the full-length transcriptome of diseased, cancerous tissues or cells, construct a comprehensive transcriptome dataset of disease/tumor samples, identify disease/tumor-specific transcripts based on grouping, and search for biomarkers for disease/tumor diagnosis and prevention.
Iso-Seq reveals complexity and diversity of alternative splicing isoforms in breast cancer transcriptome
There are widespread transcriptome alterations in tumors, but the full spectrum of splicing isoforms in cancer is not fully understood. Thousands of previously unannotated isoforms were identified in breast cancer samples, of which approximately 30% affect protein-coding exons and may alter protein localization and function. Transcription and translation of these novel isoforms were supported by extensive cross-validation with multi-omics datasets. Long-read sequencing revealed the complexity, subtype specificity, and clinical relevance of previously unknown isoforms and splicing events in breast cancer, providing a rich resource for immuno-oncology therapeutic targets.
Background
Transcriptome and proteome diversity is influenced by alternative splicing (AS), transcription initiation, and polyadenylation. Human tumors, including breast cancer, show extensive changes in splicing isoform repertoires. Short-read RNA-seq relies on alignment to a reference genome and cannot fully reveal splicing repertoires. Long-read sequencing can accurately capture full-length isoforms from beginning to end without the need for reference-based transcript reconstruction.
Methods
Sample sources: 4 normal human breasts (normal samples consisted of 2 cell lines and 2 normal breast tissues) and 26 tumor samples (breast cancer samples included 13 primary breast tumor biopsies (3 hormone-positive ER+/PR+, 3 HER2+; and 7 triple-negative breast cancers), 9 patient-derived xenograft (PDX) tumors, and 4 breast cancer cell lines).
Long-read sequencing: Single-molecule real-time (SMRT) sequencing using the PacBio RSII and Sequel platforms.
Data analysis: Sequence correction and clustering were performed using the ToFU (Transcript Isoforms Full length and Unassembled) pipeline.
Isoform classification: Isoforms were classified as known or novel based on alignment with GENCODE v.30.
Multi-omics data support: CAGE, ATAC-seq and 3'-seq data were combined to verify the transcription start and end sites of the new isoforms.
Results
The full-length transcriptome dataset of breast cancer was constructed by merging the transcriptome data of 30 samples and removing duplicate isoforms, which ultimately contained 142,514 unique full-length isoforms, covering 16,772 annotated genes and 905 unknown sites, with an average isoform length of 2.6 kb. About 2/3 of breast cancer isoforms are newly discovered transcripts, and most of the new isoforms (81%) are derived from tumor samples. About 30% of the new isoforms affect protein-coding exons and are predicted to change protein localization and function, indicating their relevance to the study of primary breast cancer. 3,059 tumor-specific alternative splicing events (AS) were discovered, of which 35 AS events in 30 genes were associated with survival in TCGA. This study used Iso-Seq to perform full-length transcriptome sequencing on the breast cancer transcriptome that was previously thought to have been well studied. It was found that the isoforms of tumor-related genes detected by Iso-Seq were much more complex than those in the known GENCODE v.30 database, which helps to discover isoform subtypes containing new tumor immune targets.
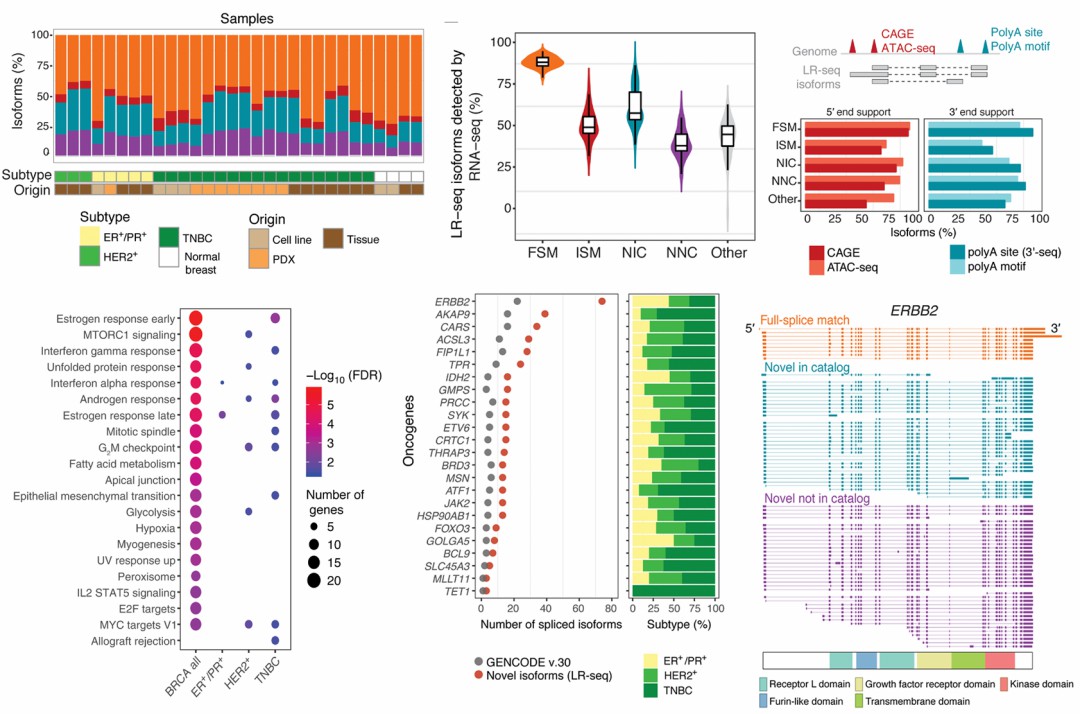 Figure 4.Isoform discovery and correlation between Isoform and tumor in breast samples.Veiga et al. 2022)
Figure 4.Isoform discovery and correlation between Isoform and tumor in breast samples.Veiga et al. 2022)
Conclusion
Iso-Seq is a long-read sequencing-based technology that can provide full-length cDNA sequences without the need for transcriptome reconstruction, thereby accurately revealing the structure and expression characteristics of transcripts. In the medical field, Iso-Seq has a wide range of applications, including exploring regional specific transcripts of tissues and cells, analyzing dynamic regulation of mRNA during development, and identifying disease-specific transcripts.
In neuroscience research, Iso-Seq has been used to analyze the expression patterns of gene isoforms in different regions of the human brain. For example, in the cerebellum, hypothalamus, and temporal cortex, studies have found a large number of previously unregistered transcript isoforms and revealed that DNA methylation may drive their expression differences. These findings are of great significance for understanding neurological diseases. In addition, during the transition from human oocytes to embryos, studies using poly(A)-inclusive RNA isoform sequencing (PAIso-seq) revealed the dynamic changes in the length of the poly(A) tail and its key role in early embryonic development.
Iso-Seq also shows great potential in cancer research. For example, in breast cancer research, the technology revealed a large number of unannotated splicing isoforms, about 30% of which affect protein-coding exons and may change protein function and localization. These new findings provide important biomarkers for cancer diagnosis and targeted therapy. In short, as an important tool for long-read sequencing, Iso-Seq provides unprecedented depth and accuracy for transcript research and disease mechanism analysis.
References
- Shimada, M., Omae, Y., Kakita, A et al. (2024). Identification of region-specific gene isoforms in the human brain using long-read transcriptome sequencing. Science advances, 10(4), eadj5279. https://doi.org/10.1126/sciadv.adj5279
- Liu, Y., Zhao, H., Shao, F et al. (2023). Remodeling of maternal mRNA through poly(A) tail orchestrates human oocyte-to-embryo transition. Nature structural & molecular biology, 30(2), 200–215. https://doi.org/10.1038/s41594-022-00908-2
- Veiga, D. F. T., Nesta, A., Zhao, Y et al. (2022). A comprehensive long-read isoform analysis platform and sequencing resource for breast cancer. Science advances, 8(3), eabg6711. https://doi.org/10.1126/sciadv.abg6711
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment