



We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Long-Read 101: Nanopore Full-Length Transcriptome Sequencing
At a glance:
- Overview
- Nanopore Sequencing vs. Next-Generation Transcriptome Sequencing
- Nanopore Sequencing: Enhancing Transcript Expression Quantification
- Nanopore Sequencing: Detection of Structural Variants
- Nanopore Sequencing: GC Content and Length Preference
Overview
Nanopore full-length transcriptome sequencing is an innovative technique that utilizes Oxford Nanopore Technologies' (ONT) sequencing platform to obtain comprehensive and uninterrupted high-quality full-length sequences of transcripts. This cutting-edge approach enables researchers to accurately identify various structural variations within transcripts, including alternative splicing, gene fusion, selective polyadenylation of alternative polyadenylation sites (APAs), allele-specific expression, and other alterations in transcript structure. Additionally, this method facilitates precise quantification of transcript expression levels, encompassing both messenger RNA (mRNA) and polyA+ long non-coding RNA (lncRNA).
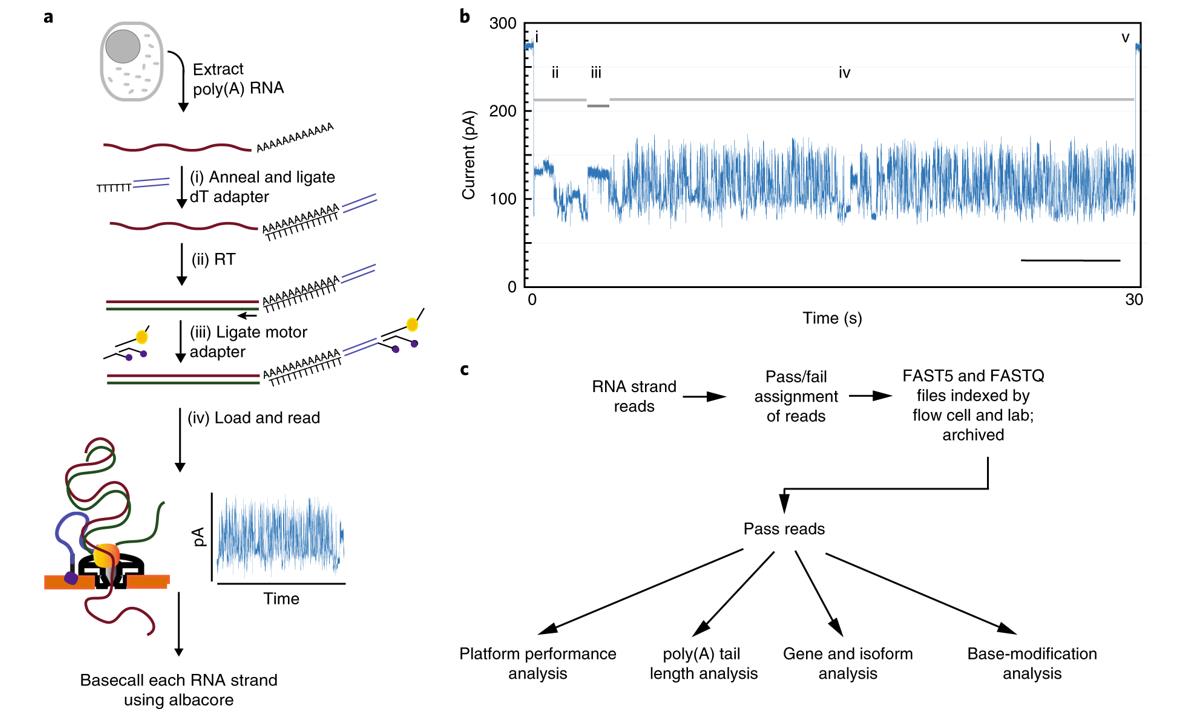 Nanopore RNA sequencing. (Workman et al., 2019)
Nanopore RNA sequencing. (Workman et al., 2019)
Nanopore Sequencing vs. Next-Generation Transcriptome Sequencing
The key differences between Nanopore full-length transcriptome sequencing and next-generation transcriptome sequencing platforms are primarily related to their sequencing technology, read length, sample processing, and data output. Here's a detailed comparison of the two approaches.
| Aspect | Nanopore Full-Length Transcriptome Sequencing | Next-Generation Transcriptome Sequencing |
| Sequencing Technology | Long-read sequencing technology based on nanopore technology. Single-stranded molecules pass through a nanopore channel, and the varying levels of obstruction caused by different bases or modifications generate distinct ionic current change signals. These signals are detected in real-time for base identification and sequencing completion. | Next-generation sequencing predominantly uses Synthesis-while-Sequencing (SDS) technology. The process involves synthesizing complementary DNA strands during sequencing, with fluorescently labeled nucleotides used to detect each base incorporation. |
| Read Length | Offers exceptionally long read lengths, with the longest reads exceeding 2 Mb. This advantage enables the capture of full-length transcripts without fragmentation, making it valuable for studying structural variations, variable splicing, and gene fusions. | Exhibits relatively shorter read lengths, typically ranging from 150 to 300 base pairs. These shorter reads necessitate RNA fragmentation and interruption during library preparation to obtain manageable read lengths, which may limit the ability to capture complete transcripts. |
| Sample Processing | Involves a simple and straightforward sample processing workflow without PCR amplification. Minimizing the involvement of PCR reduces errors and abundance changes, ensuring higher fidelity in the obtained data. | Entails a more complex sample processing phase, which includes the use of DNA polymerase, dNTPs, and bridge PCR to form clusters and amplify fluorescent signals. This complexity can increase the likelihood of introducing biases and errors during sample preparation. |
| Base Modification Information | Enables direct observation of base modifications, such as methylation (5mC, 6mA), without the need for additional bisulfite conversion or immunoprecipitation enrichment experiments. The ability to detect modifications directly enhances the understanding of epigenetic regulation. | May require additional experiments, such as bisulfite conversion or immunoprecipitation, to obtain base modification information, particularly regarding DNA methylation. These additional steps add complexity and potential variability to the workflow, impacting the overall efficiency and accuracy of detecting epigenetic modifications. |
| GC Content and Base Preference | Demonstrates lower GC content and base preference, reducing the need for RNA fragmentation and interruption during sequencing. This characteristic results in more accurate expression quantification at the transcript level. | May exhibit some bias towards GC content or specific bases, potentially affecting the accuracy of expression quantification at the transcript level. Addressing such biases may require additional data normalization and interpretation considerations. |
Nanopore Sequencing: Enhancing Transcript Expression Quantification
Nanopore full-length transcriptome sequencing presents a myriad of advantages that significantly contribute to its heightened accuracy in quantifying transcript expression levels compared to second-generation transcriptome sequencing methodologies. The following salient factors underscore the superior accuracy of Nanopore sequencing in transcript expression quantification:
- Full-Length Sequencing Capability: A paramount advantage of Nanopore sequencing lies in its inherent capacity to directly sequence full-length transcripts without the need for fragmentation and subsequent assembly. This eradicates potential splicing errors or incomplete splicing that might occur in second-generation sequencing, thereby ensuring a precise and comprehensive representation of entire transcripts.
- Precise Transcript-Level Quantification: Nanopore sequencing facilitates meticulous quantification of transcript expression at the individual transcript level. As each read spans the entire length of a transcript, it becomes more straightforward to attribute reads to specific transcripts, leading to more refined and precise transcript-level expression quantification.
- Discrimination of Transcript Isoforms: Genes often give rise to multiple transcript isoforms through alternative splicing or other mechanisms, each potentially serving distinct functions. Nanopore sequencing enables the identification and quantification of different transcript isoforms, empowering researchers to grasp the functional diversity and regulatory aspects of genes.
Nanopore Sequencing: Detection of Structural Variants
Nanopore-based full-length transcriptome sequencing surpasses second-generation methods in the precise identification of structural variants, such as alternative splicing (AS), selective polyadenylation (APA), gene fusion, and complex transcripts, due to its distinct array of advantages:
- Full-Length Sequencing: Nanopore sequencing allows for direct, unfragmented sequencing of entire transcripts. This capability facilitates the accurate delineation of complete isoform sequences, including the precise locations of transcription start sites (TSS) and transcription termination sites (TTS). Short-read NGS, by comparison, often faces challenges in this regard.
- Transcript Isoform Identification: The technology employed by Nanopore excels in determining exon connectivity with unparalleled precision, enabling the identification of intricate transcripts, involving multiple exon jumps and intronic retention. This high level of detail remains elusive with NGS.
- Accurate Fusion Gene Detection: Nanopore sequencing delivers more reliable fusion gene detection outcomes compared to next-generation sequencing. Its longer read lengths and full-length coverage offer a superior ability to pinpoint fusion events, mitigating the difficulties associated with genomic duplications and multiple comparisons.
- Selective Polyadenylation (APA) Detection: Nanopore sequencing effectively captures poly(A) tails in sequencing results, ensuring the dependable identification of APA events. Conversely, next-generation sequencing techniques often necessitate specialized polyA-seq approaches and supplementary experiments to confirm APA positions, introducing the potential for false positives and incomplete detection.
- Low Multi-Comparison Rate: The extended read lengths and reduced multiplexing rate of Nanopore sequencing contribute to a diminished multi-comparison rate when compared to short-read sequencing. This reduction in inaccuracies and biases improves the quantification of transcript levels and enhances the identification of structural variants.
- Uninterrupted Full-Length Sequences: Nanopore sequencing directly furnishes full-length transcripts spanning from the 5' to 3' end, eliminating the need for interruptions and splicing of individual transcripts. Consequently, this methodology ensures a comprehensive and accurate representation of structural variations at the transcript level."
Nanopore Sequencing: GC Content and Length Preference
Nanopore full-length transcriptome sequencing showcases distinct proclivities concerning GC content and transcript length preference, in contrast to NGS transcriptome sequencing. These variances carry profound ramifications for the precision and fidelity of gene expression quantification and structural variation analysis. Allow us to explore these disparities in greater depth:
- GC Content Preference
a) Nanopore Full-Length Transcriptome Sequencing: Harnessing the long-read capability, Nanopore sequencing exhibits a remarkable capacity to provide a more equitable representation of GC content across transcripts. Notably, the long-read datasets derived from Nanopore technology demonstrate a discernible reduction in GC preference as compared to their short-read counterparts. This advantageous attribute alleviates potential biases arising from fluctuations in GC content, thereby facilitating more accurate and robust quantification of gene expression levels. Such improvements are particularly pronounced when assessing regions with diverse GC-rich or GC-poor compositions.
b) Next-Generation Transcriptome Sequencing: In contrast, short-read sequencing technologies, typified by conventional next-generation cDNA sequencing, have shown a propensity for GC content preference. This proclivity can induce distortions in the quantification of gene expression, especially in genomic loci characterized by extreme GC content. The consequent biases may impede the accurate identification of differentially expressed genes and transcript isoforms, undermining the fidelity of the analysis.
- Length Preference
a) Nanopore Full-Length Transcriptome Sequencing: Capitalizing on its long-read capabilities, Nanopore sequencing generates datasets spanning the entirety of transcripts. As a result, the observed length bias in nanopore sequencing data is significantly ameliorated when compared to short-read datasets. This represents a substantial advantage, ensuring a comprehensive and representative coverage of transcripts with varying lengths. The technology thereby enables precise quantification of both short and long transcripts, enhancing the overall fidelity of the transcriptome analysis.
b) NGS: Short-read sequencing technologies inherently contend with a length preference bias. The necessity for transcript fragmentation to fit within limited short read lengths elevates the risk of overlooking extended transcripts or segments with substantial length. Consequently, this length bias can lead to incomplete characterization of transcript isoforms, particularly those featuring lengthy exons or intricate exon-exon junctions, thereby potentially compromising the accuracy of the analysis.
Reference
- Workman, Rachael E., et al. "Nanopore native RNA sequencing of a human poly (A) transcriptome." Nature methods 16.12 (2019): 1297-1305.
Related Services
Oxford Nanopore Sequencing Technology
Nanopore Direct RNA Sequencing
Nanopore Full-Length cDNA Sequencing
Full-Length Transcriptome Profiling Combined with Long- and Short-Read Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment