We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Long-read Sequencing for Microbiome Metagenomics
At a glance:
- Overview
- Significance of Metagenomics in Microbial Research
- The Importance of Long-read Sequencing in Metagenomics
- A Comprehensive Workflow for Long-read Metagenomic Sequencing
- Improving the Accuracy and Completeness of Long-read Metagenomic Assemblies
Overview
Metagenomics is a powerful and rapidly evolving approach for revealing uncultured microbial diversity and expanding the tree of life, as well as providing new biological insights into microorganisms inhabiting unexplored environments. Methodologically, most of the compositional and functional insights about the microbiome have been obtained based on shotgun metagenome sequencing data. The proliferation of shotgun sequencing data and advances in metagenomic approaches have greatly advanced our understanding of the diversity of microbial life. Next-generation sequencing (NGS) technologies provide huge datasets, and metagenome-assembled genomes (MAGs) have been widely used to unravel the black box of uncultured microbial majorities, providing genome-level insights. However, even high-quality MAGs can be highly fragmented (even worse in complex metageomes) when assembled using traditional short-read assays (e.g., Illumina), resulting in the loss of critical genetic information, such as ribosomal genes or mobile genetic elements.
In order to bridge the genome gap, introducing long-read sequencing generated on the Nanopore and PacBio platforms for macrogenomics can help to assemble complete genomes from complex microbial communities. CD Genomics provides comprehensive long-read metagenomics sequencing services that reveal the true diversity of complex microbiomes with extremely accurate species- and strain-level detail, which can better elucidate the mechanisms by which microbial communities function in ecosystems.
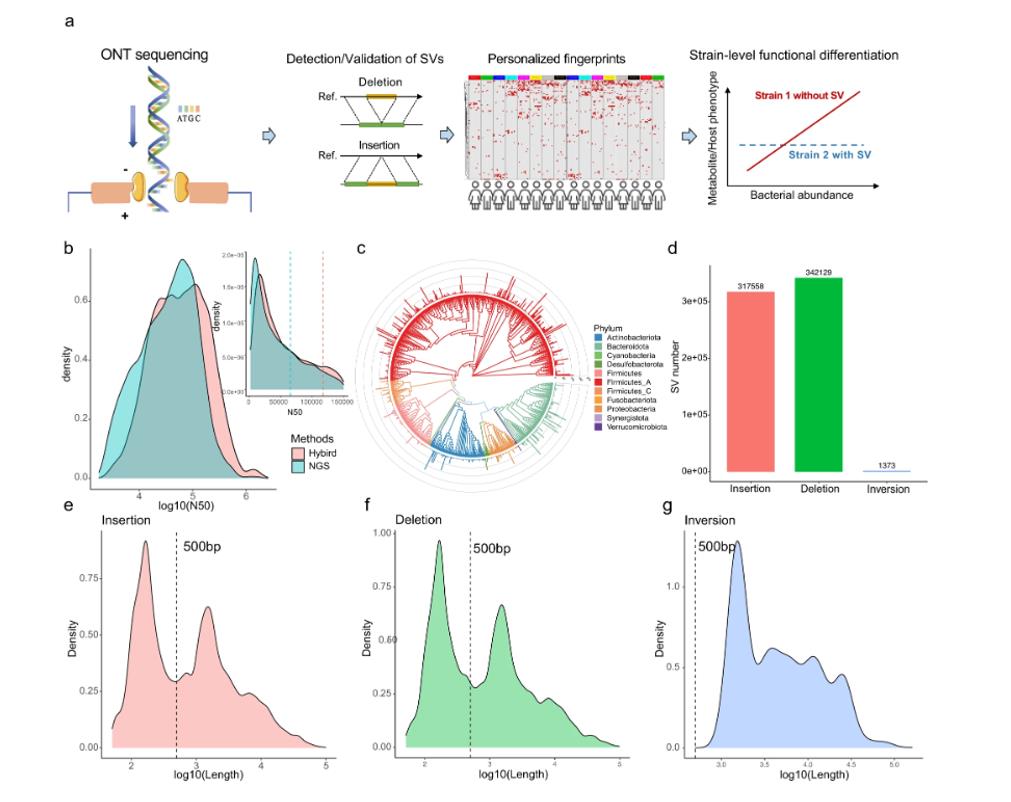 Oxford Nanopore Technology reads improved metagenomic assembly, empowered structural variations (SVs) detection and validations. (Chen et al., 2022)
Oxford Nanopore Technology reads improved metagenomic assembly, empowered structural variations (SVs) detection and validations. (Chen et al., 2022)
Significance of Metagenomics in Microbial Research
Metagenomics is a method in which entire samples are sequenced and individual community members are subsequently screened through bioinformatic analysis. Metagenomic sequencing can detect non-culturable and novel community members, and individual community member sequences can be studied to identify pathogens of difficult-to-diagnose diseases, genes that may increase virulence, and search for correlations between co-infecting pathogens that increase disease severity . The approach aims to sequence the entire genomic content of microbial samples, thereby providing more genomic information than targeted approaches.
Traditionally, microbiological research has relied on growing individual strains of microorganisms in the laboratory. However, the vast majority of environmental microorganisms are considered "unculturable" under laboratory conditions. Metagenomics circumvents this limitation, allowing researchers to delve deeper into so-called "microbial dark matter" -- the stuff that goes untapped for the most part. By decoding genetic material in environmental samples, scientists can gain insights into microbial diversity, underlying metabolic pathways, gene function and interactions in a given habitat. Therefore, this has huge implications for health, agriculture, biotechnology and environmental protection.
The Importance of Long-read Sequencing in Metagenomics
Currently, most metagenomic approaches use Illumina-based technology that produces high-precision, short reads. The short reads (150-300 bp) of Illumina sequencing make genome assembly of complex communities difficult. Short-reads are detrimental to building multiple contigs into a single scaffold for the genome, resulting in fragmented assemblies. Short-reads cannot span long repeat regions, resulting in collapse of repeat regions, providing a less complete assembly.
The PacBio and Oxford Nanopore Technologies (ONT) platforms generate significantly long-reads, allowing the bridging of repetitive genomic regions and the ability to assemble complete genomes and even plasmids. With over 99% read accuracy, these platforms are gradually setting new standards in metagenomic sequencing, as observed in recent iterations of PacBio and ONT. The application of long-read sequencing in metagenomics enables retrieval of metagenomic assembled genomes (MAGs) with high integrity. State-of-the-art strategies for long-read metagenomics use long reads to obtain a draft metagenomic assembly (ensuring maximum contiguity of the MAG), and short reads to refine and improve overall accuracy. This strategy is used to assess the human gastrointestinal microbiome, for example, in mock communities, bovine rumen, natural whey starter cultures, or wastewater.
Long-read metagenomic sequencing has the following advantages:
- Strain-level resolution: Traditional short-read sequencing may yield a metagenomic assembled genome (MAG), which is the average of multiple strains. The length and accuracy of long-read sequencing can distinguish closely related strains and capture subtle genomic variations that may be key to understanding microbial function and interactions.
- Capturing structural variants: Given their length, long-read sequencing can span large genomic regions, enabling the detection of structural variants, long repeats, and even full-length mobile genetic elements that might be missed or misassembled using short reads.
- Reduced GC bias: Traditional metagenomic workflows, especially those that rely on PCR amplification, can introduce bias targeting GC-rich regions. Long-read sequencing methods, especially those from ONTs, do not rely heavily on PCR, reducing such biases.
using the concepts written previously, rewrite this article with a high degree of complexity and specificity:
A Comprehensive Workflow for Long-read Metagenomic Sequencing
(1) Sample collection and DNA extraction: To begin with, the extraction of ultra-pure DNA from environmental samples is paramount. Given the intricate DNA input prerequisites, particularly for cutting-edge platforms like PacBio, it is of utmost importance to ensure that the DNA remains unfragmented. Employ nucleic acid stabilization solutions to preserve the integrity of DNA during transportation and storage.
(2) Library preparation: Depending on the advanced sequencing platforms, such as PacBio or ONT, the protocols for DNA library preparation vary significantly. Utilize ONT's VolTRAX V2, or similar next-generation automated sample preparation systems, to not only streamline the preparation process but also to ensure unparalleled consistency in the library.
(3) Sequencing: Upon preparing the libraries, subject them to sequencing using state-of-the-art platforms such as PacBio's Sequel II or ONT's MinION Mark II. For heightened read accuracy, it's imperative to incorporate the latest base-calling algorithms, for instance, Guppy's ultra-accurate prediction model, designed specifically for complex genomic structures.
(4) Assembly: Assemble sequences using specialized long-read assemblers such as Flye, Raven, or Redbean. Studies have shown that Flye is particularly powerful for assembling complex metagenomics, providing strain-level resolution and comprehensive plasmid assembly.
(5) Polishing: To augment the assembly precision, deploy sophisticated polishing utilities such as Medaka. Such tools capitalize on the inherent accuracy of long-read sequences to produce reconstructions that closely mirror native genomic structures.
(6) Annotation and Analysis: Leverage robust platforms like Smash Community Plus to meticulously annotate and interpret assembled genomes. This in-depth analysis provides unparalleled insights into microbial taxonomy, intricate metabolic pathways, and potential interspecies interactions.
Improving the Accuracy and Completeness of Long-read Metagenomic Assemblies
Nanopore has a low cost of entry and sequencing, enabling fast turnaround times, but nanopore sequencing has difficulty fully characterizing long homopolymeric regions and introduces insertion/deletion errors. In contrast to Nanopore sequencing, PacBio sequencing can also generate HiFi reads with high accuracy (>99%), but at the expense of read length and throughput, resulting in higher sequencing costs for metagenomic projects. These hinder the accuracy of long-read data. Three commonly used long-read specific assemblers including Flye, Raven, and Redbean can improve the completeness and accuracy of building metagenomic assemblies.
- Flye is a long-read metagenomic assembler that builds repeat maps to assemble and modify contigs. Assembly maps were then constructed using these contigs with A-Bruijn. While Flye can build more accurate metagenomic assemblies than Raven or Redbean, it also requires more time and memory.
- Raven is a fast assembler that uses an Overlay Layout Consistency (OLC) method to build assembly graphs from raw reads. For some individual assemblies, Raven can have comparable accuracy to Flye after assembly polishing, but less accurate for metagenomic assemblies.
- Redbean is another fast assembler that follows the OLC concept, using fuzzy de Bruijn graphs to build assemblies from raw reads. Redbean uses more memory than Raven but builds assemblies with less precision.
References
- Chen, Liang, et al. "Short-and long-read metagenomics expand individualized structural variations in gut microbiomes." Nature Communications. 13.1 (2022): 3175.
- Cuscó, Anna, et al. "Long-read metagenomics retrieves complete single-contig bacterial genomes from canine feces." BMC genomics. 22.1 (2021): 330.
Related Services
PacBio SMRT Sequencing Technology
Oxford Nanopore Sequencing
Long-Read Metagenomics Sequencing
Full-Length 16S/18S/ITS Amplicon Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment