We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Long-read Sequencing for Haplotype Phasing
At a glance:
- Overview
- The Importance of Haplotype Phasing
- Calculation Methods for Haplotype Phasing
- Extended Haplotype Phasing for De Novo Assembly of Long-Read Genomes Using Hi-C
- Applications of Haplotype Phasing
Overview
Humans and most other animals are diploid, meaning that each cell carries two copies of the individual genome in its nucleus, one from its mother and one from its father. Many plants have high ploidy; for example, the hexaploid California redwood has six copies of each chromosome. The number of complete sets of chromosomes (or haplotypes) in each cell is called ploidy. The number of chromosome pairs not only increases the total amount of DNA in the genome but also increases the complexity of the genome by increasing the number of alleles or alternative forms of genes. Although most of the sequences between paired chromosomes are identical, it is these differences that provide the breadth of biological variation within a species. Accurately separating and assembling these two copies or haplotypes poses a challenge to the genomics community because the two copies are very similar to each other (they come from the same species), yet are not identical. Accurate and continuous information about these small differences over extended genomic regions is needed to separate or phase these two haplotypes.
Although next-generation DNA sequencing technologies are very accurate, their utility in analyzing ambiguous and repetitive portions of the genome is limited by their short read. The advent of single-molecule long-read sequencing, such as PacBio SMRT (single-molecule real-time) sequencing, Oxford Nanopore Technologies (ONT), and Bionano Genomics, as well as high-throughput chromatin conformation capture (Hi-C) technologies, has allowed us to explore more of the genome and perform more comprehensive genome analysis. We have also been able to analyze the genomes of many other genomes, including the following Whether sequencing a giant polyploid or a diploid, the goal is to obtain a complete and accurate representation of each copy of the genome or region of interest. This is usually accomplished by assembling a haploid (single-copy) genome and then identifying the different variants and positions of the alleles.
The assembly of reference genomes is a common way to improve the utilization of genetic resources in many organisms today. However, a major challenge in assembling genomes from scratch in diploid and polyploid organisms is accurate haplotype resolution. Most reference genome assemblies collapse haplotypes that represent "mosaic" sequences, ignoring allelic variation that may be involved in important cellular and biological functions. Decompression of haplotypes into distinct sets of sequences has become a growing trend in recent genomic research because it is an important tool for addressing important clinical and biological questions such as compound heterozygosity, heterozygosity, and evolution.
The Importance of Haplotype Phasing
Researchers have been trying to improve genome assembly to fully resolve two haplotypes in the same sequencing study. The development of long-read sequencing has provided an opportunity to resolve structural variation between haplotypes that are completely absent from linear reference genomes. However, identifying variants does not provide the complete sequence of the genome. This requires phasing, or determining which variants come from the same chromosome copy ("cis") and which variants come from different copies ("trans"). One method of phasing is to use a mother-parent-child trio: variants in the child's genome that are present in only one parent must be located on the same chromosome. A second method is population inference, which infers that variants that are common in the same individual are likely to be homozygous. Both trio and population phasing are imperfect because they require additional information and can only phase certain variants. Haplotype phasing has now become a fundamental problem in the assembly of heterozygous and polyploid genomes. Haplotype phasing is the key to the most accurate representation of the genetic composition of a given organism.
Calculation Methods for Haplotype Phasing
Comparison-Based Haplotype Phasing
When a reference genome is available, the ratio-based strategy provides an efficient approach. With lower coverage sequencing, reads are mapped to that reference genome, leading to variant calling. After these variants are identified, they are concatenated into fixed-phase blocks containing a series of neighboring single nucleotide polymorphisms (SNPs). Each SNP is uniquely identified as either a reference (0) or a substitution (1). This approach relies on the plummeting cost of next-generation sequencing (NGS), and multiple algorithms have emerged, each satisfying different optimization criteria, such as minimum error correction (MEC), weighted minimum letter flip (WMLF), and maximum fragmentation cut (MFC). Recent advances have given rise to NGS-based programs such as the WhatsHap, HapCUT2, and SHAPEIT families, which are increasingly applied to complex genomes.
Assembly-Based Haplotype Phasing
Using a different approach, assembly-based phasing requires more in-depth sequencing, but in turn, it can provide more precise results covering a wide range of genomic variants, from large insertions or deletions to structural variants. The challenge lies in the presence of multiple haplotypes, which can introduce ambiguity into the initial overlapping cluster assembly. To address this problem, a common approach is to reconstruct a single representative haplotype for the entire genome. Tools such as the Redundans program and the HaploMerger pipeline have played a key role in solving these complex problems, making the assembly of heterozygous genomes more streamlined.
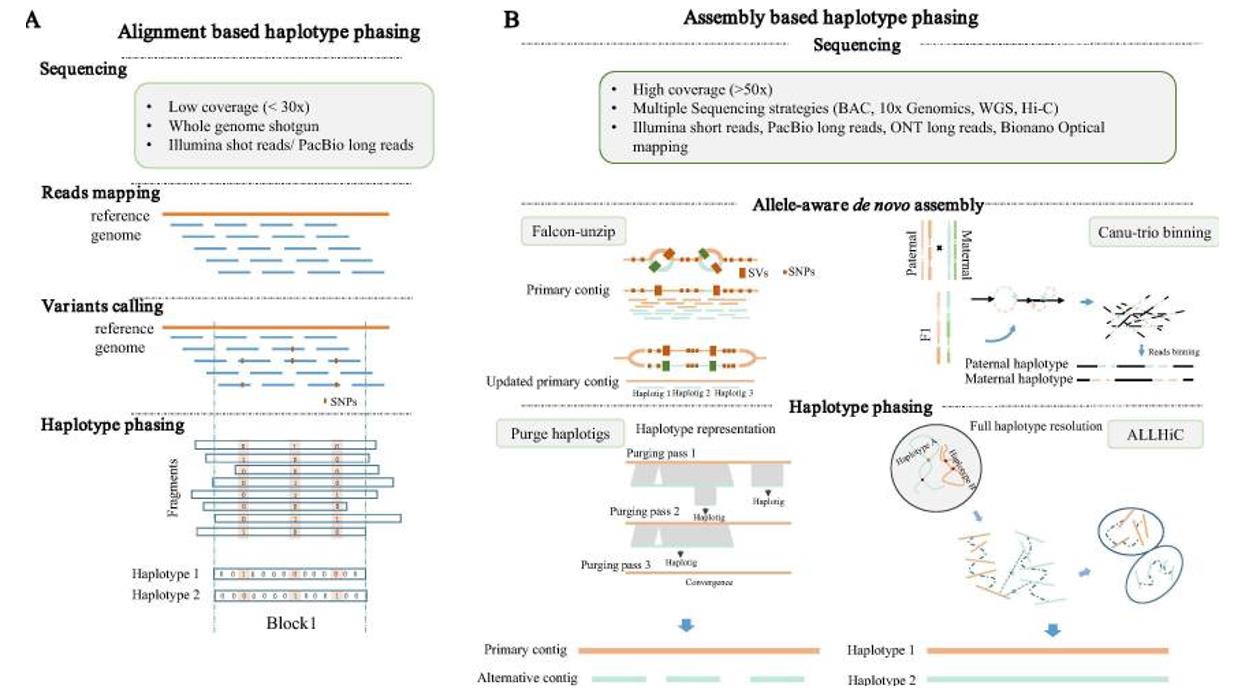 Overview of the two main classes of haplotype phasing strategies. (Zhang et al., 2020)
Overview of the two main classes of haplotype phasing strategies. (Zhang et al., 2020)
Extended Haplotype Phasing for De Novo Assembly of Long-Read Genomes Using Hi-C
Genome assembly combining PacBio long-read sequencing and neighbor-joining methods is an efficient way to construct chromosome-level genome assemblies. High-throughput chromatin conformation capture (Hi-C), a technique derived from chromatin conformation capture (3C) technology, combines chromatin neighbor-joining methods with high-throughput sequencing to obtain a fine map of chromatin interactions across a chromosome. Cross-linked chromatin is systematically cleaved and sequenced to reveal comprehensive data about sequence alignment on chromosomes.
The groundbreaking use of Hi-C is not limited to genome assembly. Over the years, it has paved the way for reliable chromosome-scale de novo genome assembly in a range of organisms from mammals to plants and insects. One of the remarkable properties of Hi-C is its ability to phase genomes onto different haplotypes at the chromosome scale, a feat made possible because homologous chromosomes occupy different regions within the nucleus. Cutting-edge tools such as FALCON-Phase and ALLHiC have been developed to take advantage of this capability and facilitate the precise phasing of genomes from simple diploids to complex polyploids.
Applications of Haplotype Phasing
Obtaining a complete picture of genetic variation
Recent advances in DNA sequencing technology and tools for assembling and phasing genomes allow the phasing of large chunks of sequence directly from a person's DNA sequencing reads. Highly accurate long reads (called HiFi reads) are particularly well suited for phasing haplotypes because they provide the high precision needed to detect single nucleotide variants (SNVs) and the length of reads that link these variants over a long range. Phasing involves separating maternal and paternal genetic copies of each chromosome into haplotypes to obtain a complete picture of the genetic variation.
Resolving variants missed by short read segments
Short-read methods miss repetitive regions, and long-read sequencing can improve pharmacogenetic testing by both characterizing repetitive variants and unambiguously phasing clinically significant variants. In summary, long-read sequencing can detect and phase alleles missed by short-read sequencing.
Studying allele-specific expression (ASE) or allelic imbalance (AI)
ASE or AI reveals the preferential expression pattern of one parental allele relative to another. This differential expression pattern may be caused by allelic variants located in cis-regulatory elements that are capable of interacting with environmental factors to regulate complex expression networks and ultimately lead to large phenotypic variations. Current methods for identifying ASEs rely on phasing RNA-seq reads to different haplotypes. Typically, RNA-seq reads generated on the Illumina short read length sequencing platform are aligned to a reference genome and variants belonging to each parental genome are phased and the corresponding read lengths are further used to quantify the gene expression of their respective alleles.
Facilitating the study of polyploid evolution and informing crop breeding
The identification of allelic variants can also facilitate the study of polyploid evolution and inform crop breeding. The complex polyploid sweetpotato genome was first assembled into a shared reference genome, and a novel haplotype phasing method successfully generated haplotype-resolved genome assemblies. Phylogenetic analyses using phased allelic variation identified a total of six haplotypes, thus tracing the history of hexaploidization in the sweetpotato lineage. In the sugarcane project, researchers developed a novel ALLHiC algorithm that is capable of constructing allele-aware and chromosome-scale assemblies of homozygous polyploid sugarcane genomes through a combination of PacBio long-read sequencing and Hi-C technologies.
Reference
- Zhang, Xingtan, et al. "Unzipping haplotypes in diploid and polyploid genomes." Computational and structural biotechnology journal 18 (2020): 66-72.
Related Services
PacBio SMRT Sequencing Technology
Oxford Nanopore Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment