We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Long-read Sequencing for Viral Genomes Study
At a glance:
- Overview
- Advantages of Long-read Sequencing in Viral Genome Research
- Practical Applications of Long-read Sequencing in Viral Genome Research
- Long-read Sequencing Tools for Viral Genomes
Overview
The importance of viruses in community composition and nutrient cycling in various ecosystems, including soil, is increasingly recognized. In addition, the COVID-19 pandemic brought a sudden urgency to virus research, prompting many to look more deeply into all the tools available to characterize viral genomes. Historically, studies of viral genomes have relied heavily on short-read sequencing. While short-read sequencing of metagenomes has provided valuable insights into the composition of many viruses, it often fails to provide a comprehensive view, especially in complex viral communities with a high degree of microdiversity.
In recent years, long-read sequencing has become increasingly popular for its advantages in assembling and resolving complex DNA regions from scratch, as well as identifying full-length RNA molecules. Long-read sequencing holds the promise of a more detailed and complete reconstruction of viral genomes, thereby broadening our understanding of their biology, evolution, and interactions with their hosts. Despite their enormous impact on human health, viruses have very small and simple genomes, some of which are only a few thousand bases in length and most of which lack any repetitive structures. Long-read sequencing has become a powerful tool to fully characterize these populations.
There are two important long-read sequencing technologies, including single-molecule real-time (SMRT) sequencing from Pacific Biosciences and nanopore sequencing from Oxford Nanopore Technologies. Although current long-read methods have lower coverage and are more error-prone than short-read sequencing, these methods continue to have advantages in identifying transcriptional isoforms, including multiple spliced RNAs and transcript-length variants, as well as overlapping transcripts and alternative multiple cis-trans RNA molecules. The advent of long-read sequencing technologies has ushered in a new era of viral genome research, revealing previously unknown complexities. CD Genomics provides comprehensive Viral Whole-Genome Resequencing and Viral Genome De Novo Sequencing Services to sequence and assemble the viral genome.
Advantages of Long-read Sequencing in Viral Genome Research
- Meta-Assembled Virus (MAV) Studies: Viral communities or viral groups often contain multiple viral strains. Conventional short-read sequencing typically integrates this diversity into the shared genome, thereby masking individual strains. However, long-read sequencing can distinguish between these strains, providing a richer picture of the virome and capturing previously unnoticed strain-level diversity.
- Complex Viral Communities: Viral populations within infected hosts are often heterogeneous. Depending on the mutation rate of the virus, populations can range from dominant variants to multiple closely related genomes called quasispecies. This genomic diversity often plays a key role in the ability of viruses to evade host defenses, spread, and resist treatment. Long-read sequencing can fully characterize these clusters, revealing their internal diversity and evolutionary trajectories.
- Large Repetitive Genomes: Giant viruses or viruses with highly repetitive genomes have traditionally been difficult to sequence using short-read methods. The continuity provided by long-read sequencing has proved advantageous in this context.
- Variation in Viral Populations: Highly accurate long-read sequencing can comprehensively characterize all variations in a viral population, not just the major variants. Often, this more detailed understanding of complex viral populations reveals important aspects of biology, including how viral infections evolve over time or in response to treatment.
Practical Applications of Long-read Sequencing in Viral Genome Research
Influenza
The long-term challenge of influenza is its ability to mutate and adapt rapidly. While a universal vaccine remains elusive, HiFi sequencing has revealed the subtle evolution of this virus. In particular, sequencing multiple samples from a patient with recurrent infections has shown the virus' adaptation to treatment. In addition, sequencing has identified minor strain variants that facilitate transmission during influenza pandemics.
Hepatitis C virus (HCV) has demonstrated the challenge of viral drug resistance. SMRT sequencing has played an important role in characterizing the evolutionary trajectory of drug-resistant HCV variants. These insights come from sequencing that identifies mutations conferring resistance even in low-abundance clones, highlighting the insidious threats lurking in the viral population.
HIV
HIV's ability to integrate into the host genome and remain latent poses unique research challenges. Long-read sequencing has been revolutionary in generating full-length viral genome sequences from protoviruses. Such insights have enabled researchers to determine which part of the latent viral reservoir may still trigger new infections. In addition, unique HIV variants in the brain associated with HIV-induced dementia were identified, underscoring tissue-specific viral evolution.
SARS-CoV-2
SARS-CoV-2 research has greatly benefited from long-read sequencing. Detailed genomic studies allow scientists to juxtapose viral evolution with host immune responses, painting a comprehensive picture of host-pathogen interactions.
Long-read Sequencing Tools for Viral Genomes
viralFlye
Based on the promise of long-read sequencing, tools such as ViralFlye have been developed to identify and analyze MAV in these assemblies. While most viral genomes may not inherently require long-read sequencing for assembly, viralFlye's ability to characterize quasispecies and other diverse groups within an infected host makes it invaluable. In addition, its ability to characterize complex viral populations in detail can help decipher the evolution of a viral infection or its response to therapy.
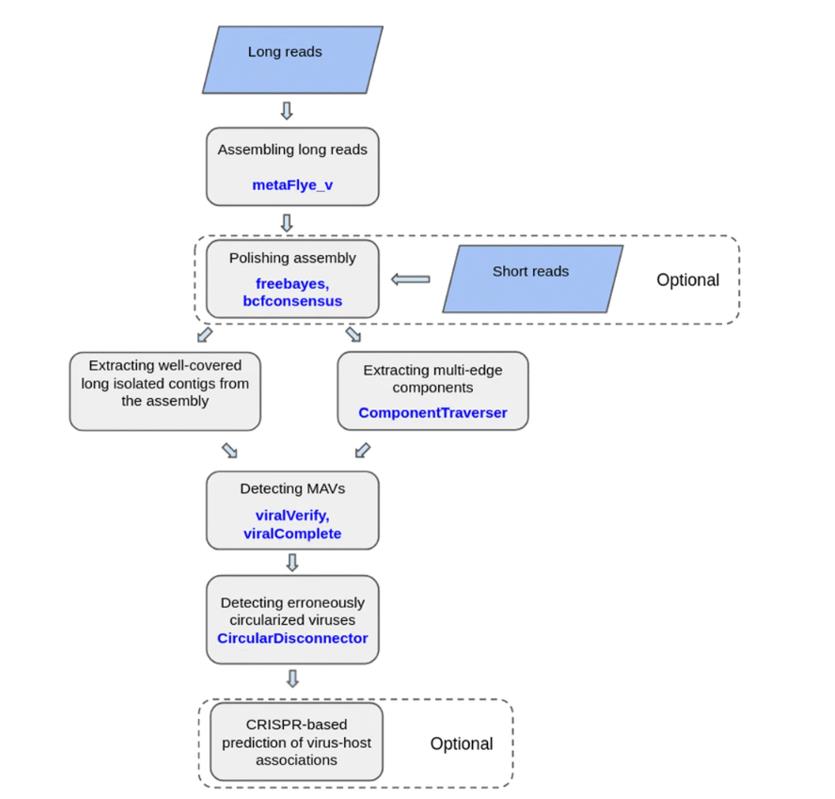 Graphical representation of the viralFlye pipeline. (Antipov et al., 2022)
Graphical representation of the viralFlye pipeline. (Antipov et al., 2022)
VirION2
The successor to the VirION method, VirION2 optimizes laboratory protocols and analytical methods to improve long-read sequencing of viral genomes. From DNA clipping to sequencing, the tool integrates short-read metagenomics to enhance results. The VirION2 method was found to recover longer, more complete viral sequences, especially compared to its predecessor. These tools not only push the boundaries of our understanding of viruses but also optimize the methodology to ensure accurate and comprehensive results.
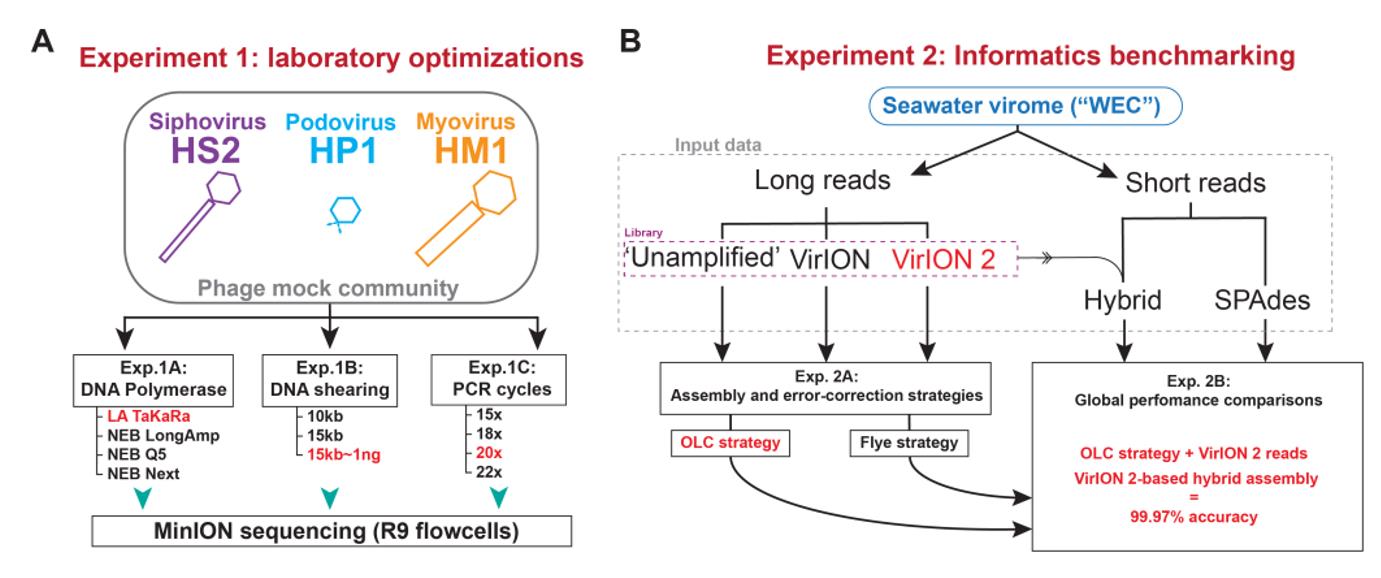 Overview of wet lab optimization experiments and informatic benchmarking. (Zablocki et al., 2021)
Overview of wet lab optimization experiments and informatic benchmarking. (Zablocki et al., 2021)
References
- Antipov, Dmitry, et al. "viralFlye: assembling viruses and identifying their hosts from long-read metagenomics data." Genome Biology. 23.1 (2022): 1-21.
- Zablocki, Olivier, et al. "VirION2: a short-and long-read sequencing and informatics workflow to study the genomic diversity of viruses in nature." PeerJ. 9 (2021): e11088.
Related Services
PacBio SMRT Sequencing Technology
Oxford Nanopore Sequencing
Viral Genome De Novo Sequencing
Viral Whole-Genome Resequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment