We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy


Tell Us Your Project
We are dedicated to providing outstanding customer service and being reachable at all times.
Maize Genome Complexity Traversed with Long-read Sequencing
At a glance:
Overview
Maize is one of the most important crops in the world and has a long history as a classical model organism in genetic research. Maize is known for its excellent chromosome cytology and rich history of transposon studies. They are diploids with 10 chromosomes, and transposons make up the bulk of the maize genome (about 85%). Their accumulation over millions of years has moved genes away from each other and separated genes from their regulatory sequences. There are also large inversions and other structural variations that contribute to fitness as well as significant changes in genome size caused by tandem repeat arrays. The maize genome is very close in size to that of humans and also contains more than 80% repetitive sequences, so it is also known as a model for complex genomes. Such rich, repetitive, and mobile sequences pose a computational challenge to accurately assemble the maize genome sequence.
A sketch of the maize genome was released in 2009, and was sequenced based on Sanger sequencing of bacterial artificial chromosomes and fosmids. due to cost and read length constraints, these early reported genome sketches typically had tens of thousands of gaps. For example, the first reported genome of the maize B73 self-inbred line had more than 100,000 gaps, and each tiled bacterial artificial chromosome sequence averaged more than a dozen gaps.
Long-read sequencing technologies such as PacBio and Oxford Nanopore have contributed greatly to improving maize genome assembly. The method generates reads tens of kilobases in length, making it suitable for improving genome continuity, closing gaps in current reference genomes, and identifying structural variation between genomes. In recent years, high-quality genome assemblies of more than 30 maize inbred lines based primarily on PacBio sequencing have been issued, such as the B73 genome, the Mo17 genome, and the entire chr3 and chr9 assemblies of the maize line B73-Ab10.
The Cases of Maize Genome Assembly Using Long-read Sequencing
Maize Genome Assembly by PacBio Long-read Using Canu version 1.8
PacBio long-read typically has a relatively high error rate compared to Illumina short-read. In addition, a large proportion of sequencing errors tend to be randomly distributed, which can be corrected by increasing sequencing coverage or by polishing the assembled Illumina short read lengths with higher accuracy. Today, several assembly tools have been designed for PacBio long-read, including Canu, Falcon, Falcon, and WTDBG2.
The researchers used Canu version 1.8 for maize genome assembly by PacBio long-read sequencing. For large and complex plant genomes, Canu-generated assemblies are often fragmented, and additional scaffolding methods are required to improve genome assembly. The BioNano Optical Atlas significantly improved genome assembly and increased N50 from 0.39Mb to 120.9Mb by anchoring 15,550 PacBio contigs into 29 super-brackets and 8486 unbricked contigs. Scaffolded using Dovetail Hi-C mapping, the final assembled genome was 2.29 Gb in length, containing 10 pseudochromosomes with a total length of 2.11Gb, and 8440 unassigned contigs with a total length of 177.23Mb. Therefore, PacBio SMRT long-read sequencing, combined with BioNano optical mapping and Dovetail Hi-C mapping technologies, can help researchers assemble high-quality maize reference genomes.
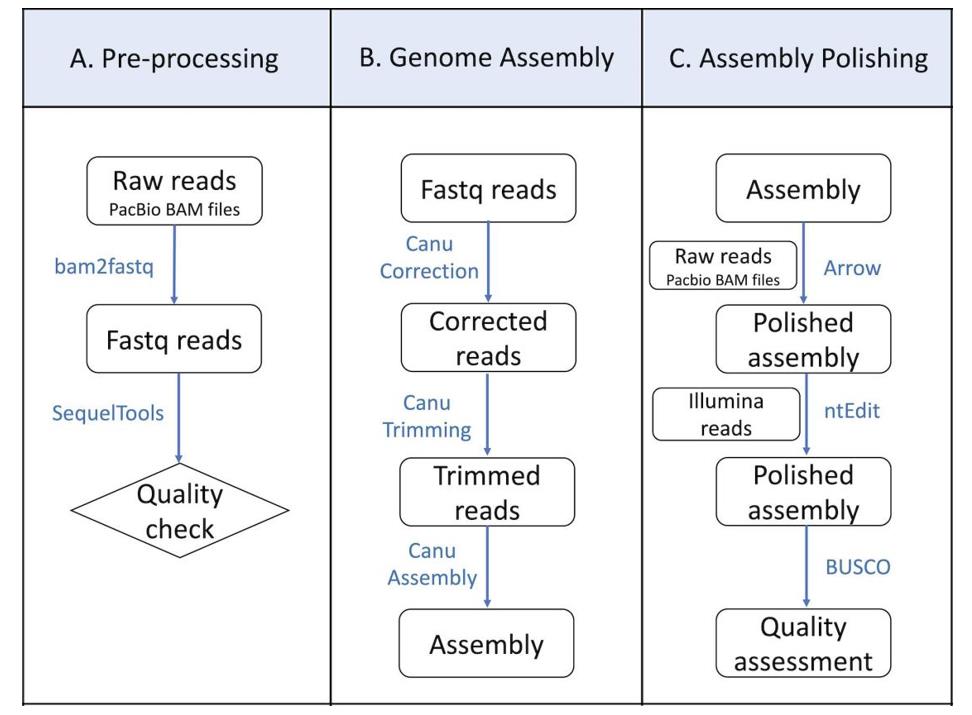 Flowchart showing various steps for pre-processing of the PacBio raw reads, genome assembly, and polishing. (Hu et al., 2022)
Flowchart showing various steps for pre-processing of the PacBio raw reads, genome assembly, and polishing. (Hu et al., 2022)
Gapless Assembly of Maize Chromosomes Using Long-read Technology
The most challenging genomic region to assemble is the tandem repeat array, which exceeds the read length of current sequencing technologies. Maize contains 156bp of mitotic repeats, 9,349bp of 45S rDNA repeats, and 341bp of 5S rDNA repeats. In addition, maize contains two types of abundant knob repeat sequences found on chromosome arms, the major knob 180 repeats (180bp) and the minor TR-1 repeat. Knob repeats occur in arrays extending over tens of megabases and pose a major obstacle to genome-wide assembly.
Creating gapless telomere-to-telomere assemblies of complex genomes is one of the ultimate challenges of genomics. Researchers used two independent assemblies and an optical mapping-based merging pipeline to generate a maize genome (B73-Ab10) consisting of 63 overlapping clusters and 162Mb of overlapping cluster N50. This approach successfully created a gapless telomere-to-telomere assembly of the complex maize genome and significantly improved the continuity of the entire genome, including the kinetochore and knob regions.
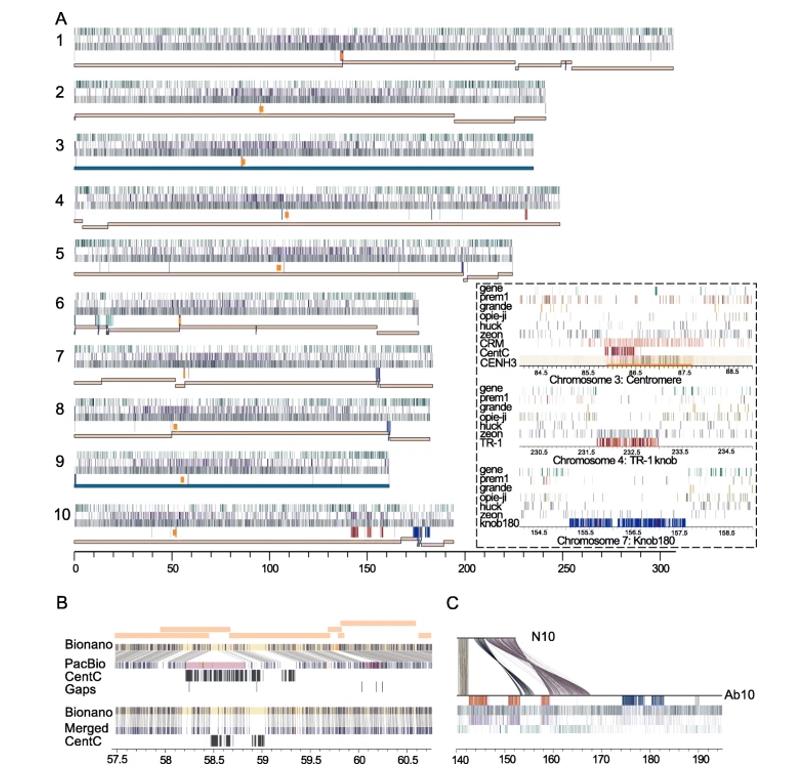 Assembly of the B73-Ab10 genome. (Liu et al., 2020)
Assembly of the B73-Ab10 genome. (Liu et al., 2020)
Complete Telomere-to-Telomere Assembly of the Maize Genome
The maize inbred lines B73 and Mo17 are among the best performing early commercial hybrids and the most widely used parental lines for biparental genetic mapping populations. Therefore, the generation of their genome sequences is of great importance. Researchers successfully obtained a complete genome assembly of maize with each chromosome entirely traversed in a single contig by generating depth-covered ultra-long Oxford nanopore technology and PacBio HiFi reads. The 2,178.6Mb T2T Mo17 genome sequence was obtained from a single contig. The 2,178.6Mb T2T Mo17 genome with a base accuracy of over 99.99% unveiled the structural features of all repetitive regions of the genome.
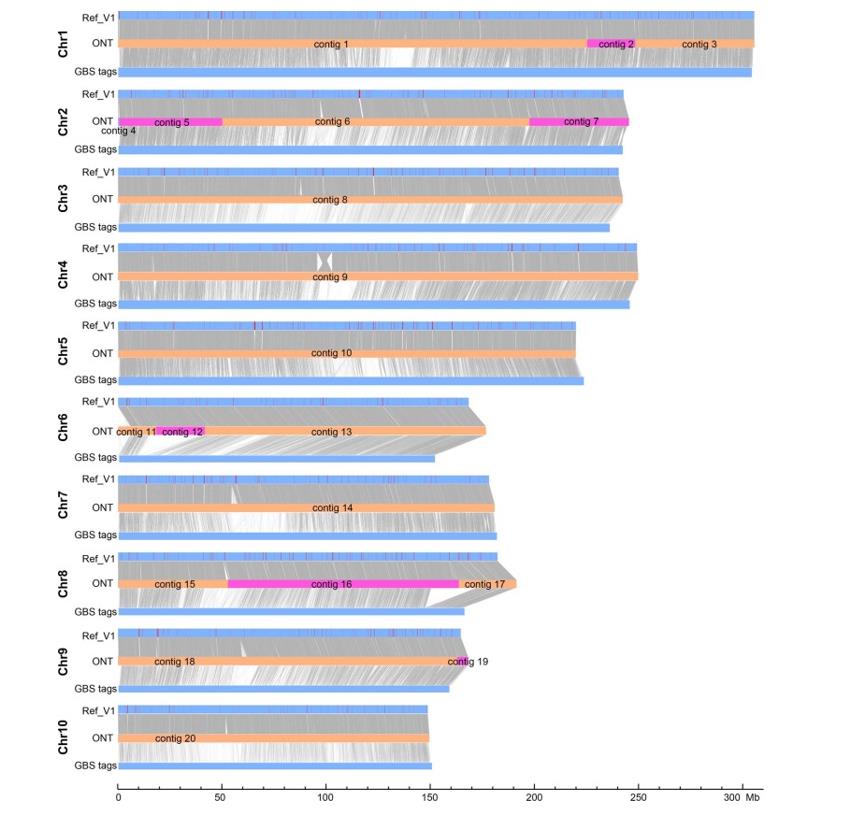 Alignment of raw ONT assembly contigs with the pseudomolecules of the Mo17ref_V1 and the GBS tags. (Chen et al., 2023)
Alignment of raw ONT assembly contigs with the pseudomolecules of the Mo17ref_V1 and the GBS tags. (Chen et al., 2023)
References
- Hu, Y. and Resende Jr., M. F. R. (2022). Maize Genome Assembly with PacBio Reads. Bio-101: e4456.
- Liu, Jianing, et al. "Gapless assembly of maize chromosomes using long-read technologies." Genome Biology. 21.1 (2020): 1-17.
- Chen, Jian, et al. "A complete telomere-to-telomere assembly of the maize genome." Nature Genetics. (2023): 1-11.
Related Services
PacBio SMRT Sequencing Technology
Oxford Nanopore Sequencing
Animal/Plant Whole Genome De Novo Sequencing
For Research Use Only. Not for use in diagnostic
procedures.
Talk about your projects
For research purposes only, not intended for personal diagnosis, clinical testing, or health assessment