
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

High-Throughput Sequencing (HTS), colloquially referred to as Next-Generation Sequencing (NGS), embodies the groundbreaking technological advancement revolutionizing the discipline of genomics. This innovative approach enables researchers to sequence DNA and RNA molecules expeditiously on an unprecedented scale. This stands in stark contrast to the traditional Sanger sequencing predicated on the chain-termination method, which has been considerably restricted by its minimal throughput and substantial cost.
In lieu of this, High-Throughput Sequencing methodologies leverage the potential of parallel processing, capable of managing millions of DNA fragments simultaneously. This promotes a swift, economical sequencing of entire genomes or transcriptomes.
Such HTS platforms capitalize on a series of sequencing-by-synthesis or sequencing-by-ligation techniques for the elucidation of DNA or RNA sequences. Central to these techniques is the fracturing of DNA or RNA molecules into diminutive fragments, to which adapters or primers are subsequently affixed. These fragments undergo amplification, culminating in the generation of clusters composed of homologous sequences on a solid support structure.
The sequencing procedure encompasses the iterative determination of the nucleotide sequence within each cluster, accomplished via the detection of the integration of luminescently tagged nucleotides or cleavage of luminescent markers.
The preeminent precision and accuracy of HTS supersede that of conventional sequencing methods. They afford sequencing data of impeccable quality and minimal errors, equipping researchers with highly reliable results. This precision gains pivotal importance in fields like variant calling and mutation detection, where the exact detection of genetic alterations is indispensable for decrypting disease processes and designing bespoke therapies.
The scalability feature of HTS permits large volume sequencing of DNA or RNA within one single experimental setup. This attribute is pivotal for projects necessitating deep sequencing coverage including comprehensive genome sequencing or complex transcriptomic studies using RNA-seq. Employing high sample throughput, HTS facilitates exhaustive analysis of genetic heterogeneity, gene expression profiling, and regulatory networks across an array of biological systems.
Moreover, HTS imparts a significant advantage in speed and efficiency over traditional sequencing techniques. With the ability to generate mammoth volumes of sequencing data in a fraction of the usual time, it empowers researchers to fast-track experiments and expedite the generation of scientific insights. This, in turn, has proven transformative in clinical diagnostics where rapid mutation detection is essential in disease management.
HTS techniques are equipped with a high degree of versatility and are applicable to a wide gamut of genomic and transcriptomic analyses. Their usage spans from whole-genome and exome sequencing to ChIP-seq and RNA-seq studies. This extensive utility of HTS techniques has spurred groundbreaking advances across multiple disciplines including cancer genomics, developmental biology, and microbiology, thereby enriching our understanding of life at the molecular level.
Illumina sequencing, a groundbreaking technology in High-Throughput Sequencing, has significantly propelled the field of genomic research forward due to its unmatched scalability, precision, and cost-effectiveness. This high-performance technique, formally termed as 'sequencing-by-synthesis', encompasses the fragmentation of DNA samples, the addition of sequencing adapters, and their subsequent amplification via Polymerase Chain Reaction (PCR).
Upon completion, these augmented fragments are sequenced concurrently in a massively parallel manner, with each integrating base detected by corresponding fluorescent signals. Remarkably, Illumina sequencers possess the capacity to concurrently sequence millions of these DNA fragments, thereby rendering it an ideal tool for diverse applications, such as whole-genome sequencing, exome sequencing, and sequence analysis of RNA or RNA-seq.
Given its remarkable reliability and flexibility, Illumina sequencing has emerged as the preferred technique across a broad scope of genomic studies, ranging from foundational research to clinical diagnostics. Consequently, this method has fundamentally altered the landscape of genomics and continues to hold great promise in the actualization of precision medicine.
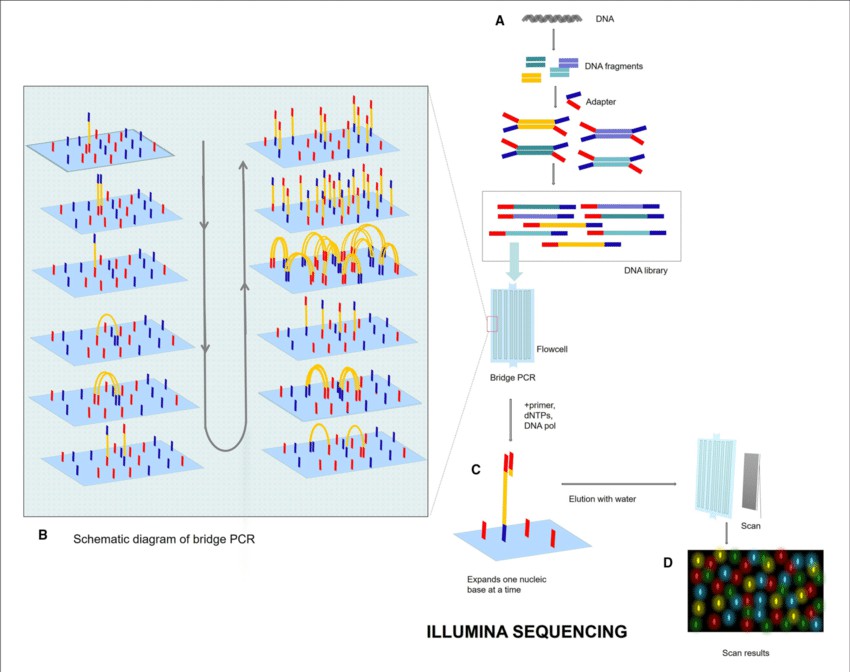 Illumina sequencing process (Lu Zhang et al,. 2021)
Illumina sequencing process (Lu Zhang et al,. 2021)
In contrast, Oxford Nanopore sequencing propels a paradigm shift by offering real-time sequencing capabilities with extended read lengths, effectively reshaping the dynamics of genomics research. This contemporary method hinges on the premise of transporting DNA or RNA molecules through a series of nanoporous structures embedded within a membrane. Perturbations in the electrical current, observed as these molecules negotiate the nanopores, are recorded and interpreted into DNA sequence data. Characterised by its swift, mobile, and direct sequencing of genetic material, this technology dispenses with the requirement for pre-amplification or fragmentation. Oxford Nanopore sequencing has proved to be transformative in the field of genomics, especially in niches requiring long-read sequencing such as de novo genome assembly, detection of structural variants, and real-time surveillance of pathogens. By incorporating a versatile and accessible design, Oxford Nanopore sequencing has broadened the horizons for genomic exploration, thus equipping researchers with a dynamic tool capable of sequencing DNA or RNA at their convenience and location of choice.
You may interested in
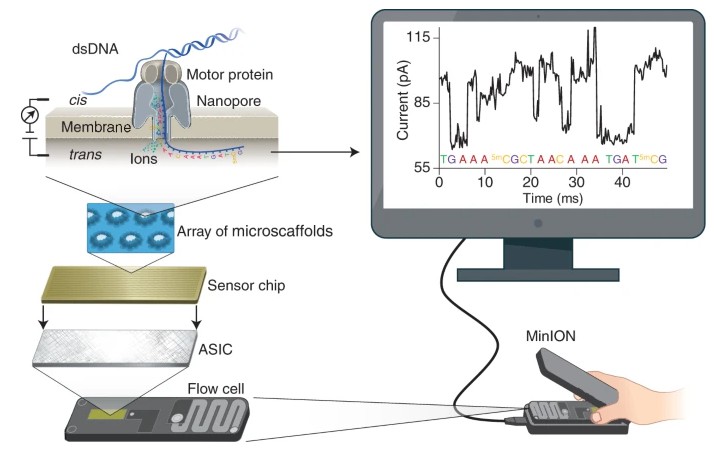 Principle of nanopore sequencing Yunhao (Wang et al, 2021)
Principle of nanopore sequencing Yunhao (Wang et al, 2021)
Pacific Biosciences (PacBio) sequencing, grounded on the foundation of Single-Molecule Real-Time (SMRT) technology, provides the scientific community with long-read sequencing capabilities combined with considerable accuracy. Diverging from Illumina and Oxford Nanopore sequencing methodologies, PacBio sequencing witnesses DNA synthesis directly in real-time, harnessing the application of fluorescently labelled nucleotides. This inventive approach facilitates the sequencing of extended DNA fragments, thereby enabling the identification of structural variants, intricate genomic rearrangements, and epigenetic modifications. Such technique has seen vast application in areas such as genome assembly, metagenomics, and transcriptomics, where the capacity to generate long reads is pivotal for deciphering complex genomic regions and capturing full-length transcripts. While PacBio sequencing may often present with lower throughput compared to Illumina sequencing, its competence in producing long, high-fidelity reads frames it as an invaluable resource for specific genomic analyses, thereby complementing the strengths of alternative sequencing platforms.
You may interested in
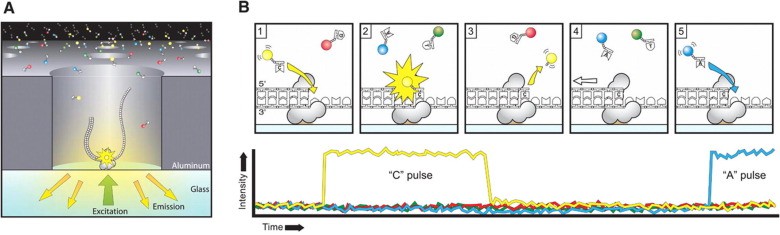 PacBio Sequencing Anthony Rhoads (et al, 2015)
PacBio Sequencing Anthony Rhoads (et al, 2015)
Ion Torrent sequencing, a brainchild of Life Technologies (currently assimilated with Thermo Fisher Scientific), employs semiconductor technology for DNA sequencing. This distinctive method detects hydrogen ions released during the process of nucleotide integration, facilitating real-time surveillance of DNA synthesis. Ion Torrent sequencers proffer swift turnaround periods and modifiable output capacity, rendering them apt for a broad spectrum of applications counting, but not limited to, focused amplicon sequencing, microbial genomic analysis, and carcinogenic mutation characterization. Although the read lengths offered by Ion Torrent sequencing might not surpass those of competitor platforms, its rapidity and elementary operational procedure make it an indispensable instrument for habitual sequencing activities in research laboratories as well as clinical environments. The ease of operation coupled with cost-effectiveness of Ion Torrent sequencing, underscores its wide-spread acceptance across manifold genomic research disciplines, further enriching the spectrum of high-throughput sequencing technologies.
Comparative overview of high-throughput sequencing technologies
| High-Throughput Sequencing Technology | Illumina Sequencing | Oxford Nanopore Sequencing | Pacific Biosciences Sequencing | Ion Torrent Sequencing |
| Principle | Sequencing-by-synthesis | Nanopore-based | Single-Molecule Real-Time (SMRT) | Semiconductor-based |
| Read Length | Short to medium | Long | Long | Short to medium |
| Accuracy | High | Variable | High | Moderate to high |
| Throughput | High | Moderate to high | Moderate | Moderate to high |
| Fragmentation Requirement | Yes | No | No | Yes |
| Real-Time Sequencing | No | Yes | Yes | Yes |
| Portability | Limited | Yes | No | Limited |
| Applications | Whole-genome sequencing, exome sequencing, RNA-seq | De novo genome assembly, structural variant detection, real-time pathogen surveillance | Genome assembly, metagenomics, transcriptomics | Targeted amplicon sequencing, microbial genomics, cancer mutation profiling |
Transcriptomics, an insightful examination of RNA transcripts engendered by the genome, has experienced tremendous advancements owing to the advent of high-throughput sequencing (HTS) technologies. The sequencing of RNA molecules, extracted meticulously from specific cells or tissues, enables researchers to unravel patterns of gene expression, instances of alternative splicing, as well as post-transcriptional modifications. With its broad-spectrum applications spanning across multiple specialties, HTS-based transcriptomics finds its utility in gene expression profiling, the discovery of non-coding RNAs, and the detailing of RNA modifications.
Gene Expression Profiling:
One of the rudimentary yet critical applications of HTS in the realm of transcriptomics is profiling gene expression, a process involving the quantification of the RNA transcript abundance within a biological specimen. Undertaking sequencing of the transcriptome enables researchers to pinpoint differentially expressed genes during variable experimental conditions, clinical states, or developmental phases. The gleaned information is indispensable in the comprehension of cellular mechanisms, signaling pathways, and the regulatory networks that underpin biological phenomena. For instance, RNA-seq-based gene expression profiling has been advantageous in scrutinizing aspects of cancer progression, development, therapeutic interventions, thus unveiling potential biomarkers and therapeutic targets.
Identification of Non-Coding RNAs:
The advent of HTS technologies has been vital in uncovering and understanding non-coding RNAs (ncRNAs), which hold significant sway in gene regulation and cellular mechanisms. Undertaking sequencing of the transcriptome enables the identification of diverse ncRNAs subclasses, including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs). These ncRNAs influence various biological roles like chromatin remodeling, RNA splicing, and post-transcriptional gene regulation. To demonstrate, miRNAs have been implicated in varying facets of cancer development and progression, thereby influencing the expression of genes involved in cellular proliferation, apoptosis, and metastasis.
Characterization of RNA Modifications:
Further, HTS-based transcriptomics also facilitates the study of RNA modifications like m6A methylation, pseudouridylation, and RNA editing, which play instrumental roles in dictating RNA stability, translation efficiency, and protein-RNA interactions. Sequencing RNA molecules with high precision allows researchers to map and quantify RNA modifications across the whole transcriptome. Such garnered information enriches our understanding of gene expression's dynamic regulation and the functional repercussions of RNA modifications in health and disease. Evidence suggests that dysregulation of m6A methylation is implicated in a myriad of human diseases, including cancer, neurodevelopmental disorders, and metabolic diseases.
You may interested in
The advent of High-Throughput Sequencing technologies has catalyzed a profound evolution in the realm of genomics research; facilitating the capacity to scrutinize genomes, transcriptomes, and epigenomes with unprecedented detail and exactitude. As the capacity of HTS has unfolded, it has burgeoned across multiple scientific frontiers, finding substantial footholds in the domains of cancer genomics, clinical diagnostics, environmental genomics, and personal genomics.
Cancer Genomics:
Arguably one of the most monumental applications of HTS resides within the sphere of cancer genomics. It has fundamentally recalibrated our perception of the underlying molecular architecture within tumors and engendered the pioneering field of precision oncology. Through sequencing both the genomic and transcriptomic fabric of these rogue cells, scientists can excavate key information including driver mutations, functionality of oncogenic paths, and crucial therapeutic targets (Schwaederlé et al., 2015). A case in point is the identification of highly specific mutations in pivotal genes such as EGFR, BRAF, and ALK which have steered the creation of targeted therapeutics revolutionizing the clinical outcome for patients with specific cancer types (Hyman et al., 2017). Furthermore, implementing HTS in a non-invasive manner as liquid biopsies provides robust surveillance of tumor progression and response to treatment, offering valuable intel on disease advancement and strategic personal treatment approach (Diaz and Bardelli, 2014).
Clinical Diagnostics:
In the realm of clinical diagnostics, HTS has emerged as a game-changing tool, providing a comprehensive diagnosis and customized treatment protocol. By sequencing patient-centric exomes or genomes, clinicians can decode the existence of pathogenic mutations, genetic predispositions, and pharmacogenomic markers instrumental in qualifying treatment responses (Yang et al., 2013). This HTS-guided approach has proven particularly influential in diagnosing rare genetic disorders, where conventional diagnostics often stumble upon inconclusive results (Bick et al., 2017).
Environmental Genomics:
HTS has robust applications reaching beyond clinical settings. In environmental genomics, it has provided a novel perspective to study microbial communities, biodiversity, and ecosystem dynamics. By sequencing environmental samples through metagenomic methodologies, researchers can unravel the mysteries of microbial diversity and their role in biogeochemical cycles (Tringe and Hugenholtz, 2008). Enhanced understanding of gene expression dynamics through transcriptomics and metatranscriptomics informs effective ecosystem management and conservation strategies.
Personal Genomics:
In personal genomics, the utility of HTS technologies permeates through, enabling individuals to gain insights into their genetic proclivities, genealogies, and health risks. It allows direct-to-consumer genetic testing services to offer individuals access to personalized genomic information for ancestry mapping, trait analysis, and disease predisposition prediction (Tandy-Connor et al., 2018). Furthermore, HST has significant applications in pharmacogenomics by identifying genetic variants that affect drug metabolism and treatment responses, paving the way for personalized drug regimens tailored to an individual's unique genotype (Caudle et al., 2014). These advancements underscore the profound and transformative impact of HTS in genomic research, propelling breakthroughs in the spheres of medicine, ecology, and personalized healthcare.
You may interested in
Epigenomics, encapsulating the intricacies of epigenetic alteration impact on gene expression and cellular phenotypes, stands as a crucial discipline interfacing biology and medicine. The inception of high-throughput sequencing technologies has catalyzed a paradigm shift in epigenomic research, enabling an exhaustive analysis of epigenetic signatures scattered across the genome. The applications of HTS are multipartite, streamlining the mapping of DNA methylation, histone modification patterns, chromatin accessibility, and presenting an insight into three-dimensional chromatin architectures.
DNA Methylation Profiling:
DNA methylation, distinguished by the addition of methyl groups to cytosine bases, is integral to gene expression regulation, buttressing genomic stability, and assisting cellular differentiation. HTS-oriented methods such as bisulfite sequencing and the reduced representation bisulfite sequencing (RRBS) have propelled genome-wide DNA methylation profiling into a realm of single-nucleotide resolution (Cokus et al., 2008). Bisulfite-treated DNA sequencing helps disclose methylated and unmethylated cytosines thereby unveiling DNA methylation patterns pervading the genome. Such knowledge is monumental to comprehend the significance of DNA methylation in normative development, aging, along with vulnerabilities to disease conditions like cancer and neurological impairments (Meissner et al., 2008).
Histone Modification Mapping:
Histone modifications, encompassing acetylation, methylation, phosphorylation, and ubiquitination, are cardinal to chromatin structuring, gene regulation, and genome organization. HTS-integrated methods, such as chromatin immunoprecipitation succeeded by sequencing (ChIP-seq), have revolutionized the charting of histone modifications across the genome (Barski et al., 2007). Sequencing DNA fragments enriched for specific histone modifications facilitates the identification of genomic regions linked with active or repressive chromatin states. This knowledge is pivotal to shed light on regulatory mechanisms driving gene expression, enhancer activity, and chromatin dynamics across health and disease spectrum (Heintzman et al., 2007).
Chromatin Accessibility Assays:
Chromatin accessibility, an indicator of DNA's amenability to regulatory proteins and transcription factors, is central to gene expression and regulatory element functionality. HTS-driven assays such as the assay for transposase-accessible chromatin using sequencing (ATAC-seq) and DNase-seq, allow high-resolution, genome-wide profiling of chromatin accessibility (Buenrostro et al., 2013). Accessible chromatin region sequencing helps pinpoint active regulatory factors including promoters, enhancers, and insulators, and expose their roles in gene regulation and cellular identity. Such insights prove critical to deconstruct the epigenetic foundation of cellular differentiation, tissue evolution, and disease etiology (Song and Crawford, 2010).
Three-Dimensional Chromatin Architecture:
The latest strides in HTS technologies have also eased the exploration of three-dimensional chromatin architecture and genomic organization. Protocols like Hi-C, chromatin interaction analysis by paired-end tag sequencing (ChIA-PET), and HiChIP equip researchers to probe the spatial genome organization and discern chromatin loop and domain formations (Lieberman-Aiden et al., 2009; Fullwood et al., 2009). Sequencing chromatin interactions facilitates the reconstruction three-dimensional genome models and discernment of long-range interactions between regulatory factors and target genes. This information is pivotal to comprehend the superior chromatin structure, genome folding principles, and the spatial genome organization within the nucleus (Rao et al., 2014).
You may interested in
Research into the microbiome, a focus on ubiquitous microbial communities across diverse ecosystems, has witnessed significant advancements with the advent of High-Throughput Sequencing technologies. This critique foregrounds the central role of HTS in elucidating microbiome composition and function, highlighting applications in exploring microbial diversity, discerning community structures, effecting functional profiling, and elucidating the complex microbiome-host associations.
Characterizing Microbial Diversity:
HTS has emerged as a game-changing approach to demystifying microbial diversity, offering an exhaustive portrait of microbial communities across various ecosystems such as the human gut, soil, ocean, and atmospheric milieux. Through techniques like amplicon sequencing of the 16S ribosomal RNA gene and whole-genome shotgun metagenomic sequencing, the taxonomic composition and abundance of microbial constituents within samples become discernable (Turnbaugh et al., 2007). By facilitating parallel analysis of millions of DNA sequences, HTS empowers researchers to identify elusive and relatively scarce microbial species, decipher community dynamics, and evaluate the influence of environmental variables on microbial diversity.
Deciphering Community Structure:
Beyond taxonomic profiling, HTS holds considerable potential in elucidating microbial community architecture and composition. Producing intricate data on microbial populations, HTS enables the recognition of community shifts, ecological linkages, and keystone species within intricate microbial ecosystems. Approaches such as metagenomic sequencing deliver robust evidence of microbial communities' functional potential, offering insights into their involvement in nutrient cycling, bioremediation, and disease propagation (Gilbert et al., 2010). The evolution of single-cell sequencing technique has further unveiled the genomic diversity, metabolic capacities, and interactive networks of individual microbial cells within a community.
Functional Profiling and Metagenomics:
The deployment of HTS in metagenomic sequencing facilitates exploration of microbial communities' functional potential through collective genetic content examination in a sample. Metagenomic investigations reveal the nuances of microbial metabolism, gene functionalities, and pathways implicated in environmental operations and host-microbiome interplay (Qin et al., 2010). By annotating genes and predicting metabolic pathways, metagenomics enables identification of microbial traits relevant to biogeochemical cycling, antibiotic resilience, and host wellbeing. Complementary to metagenomics are metatranscriptomic and metaproteomic methods, which uncover active gene transcription and protein profiles of microbial communities, respectively, thereby providing dynamic insights into functional activities.
Microbiome-Host Interactions:
HTS underpins a nuanced understanding of microbiome-host interactions and their bearing on human health and disease manifestation. Investigations powered by HTS have shed light on the gut microbiome's role in modulating host metabolism, the immune response, and vulnerability to an array of conditions including obesity, inflammatory bowel diseases, and metabolic syndromes (Arumugam et al., 2011). The integration of multi-omic data from microbiome and host samples enables researchers to decode the intricate reciprocity between microbial communities and host physiological response, potentially enabling personalized medicine strategies targeting the microbiome.
You may interested in
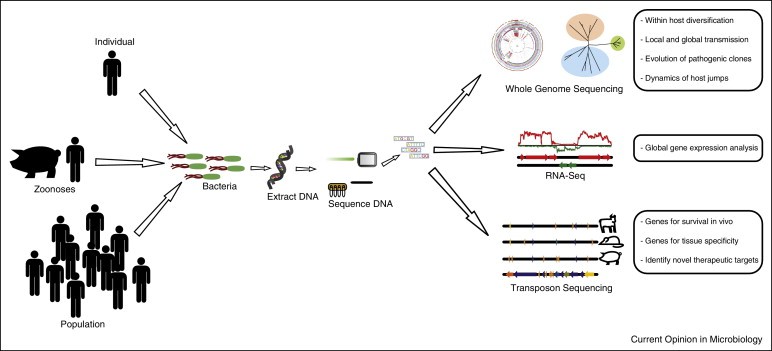 Schematic diagram summarizing the applications of high throughput sequencing for studies of the epidemiology, evolution and pathogenesis of bacterial infections. (Paul R McAdam et al, 2014)
Schematic diagram summarizing the applications of high throughput sequencing for studies of the epidemiology, evolution and pathogenesis of bacterial infections. (Paul R McAdam et al, 2014)
The initial phase in High-Throughput Sequencing necessitates creating a library in which DNA or RNA samples undergo fragmentation, are marked with sequencing adapters and are subsequently amplified, resulting in the production of sequencing libraries. Although each application's specific requirements and the sequencing platform employed may slightly alter this process, the standard approach involves enzymatic reactions, purification processes, and the evaluation of quality control to ensure the resulting sequencing libraries possess integrity and reliability.
Following the successful preparation of the sequencing libraries, the next course of action involves loading these prepared libraries onto the sequencing platform. In this phase, a sequence of nucleotide bases is progressively integrated into the formation of growing DNA strands. With each individual incorporation of a base, the sequencing instrument records and identifies it. The sequencing platforms employed in high-throughput procedures generate an enormous quantity of raw sequencing data. This raw data necessitates subsequent processing and analytical examination to extract biologically meaningful information.
High-Throughput Sequencing underscores the crucial importance of data analysis, which involves the handling, alignment and interpretation of generated sequencing data. Bioinformatics pipelines are utilised to trim raw reads, correspond them to reference genomes or transcriptomes, and discern genetic variations or patterns of differential gene expression. Owing to advanced computational mechanisms and algorithms, researchers are empowered to obtain biological insights from large-scale sequencing datasets, thereby facilitating novel discoveries in diverse research areas such as genetic basis of diseases, evolutionary biology and personalised medicine.
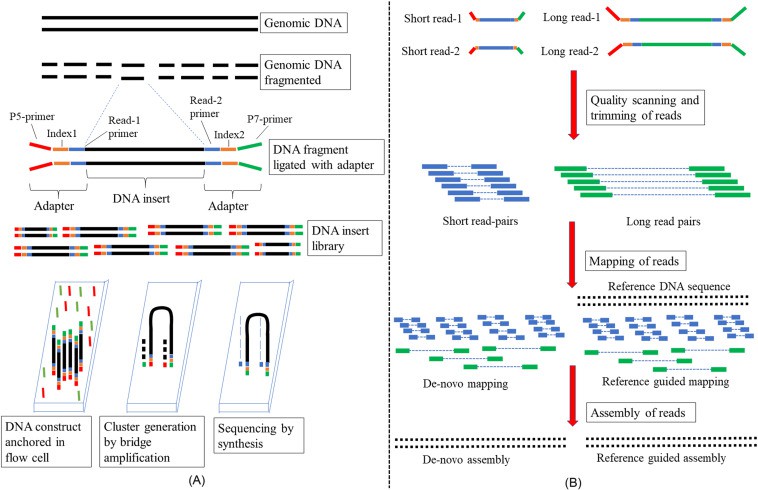 High-throughput DNA sequencing.
High-throughput DNA sequencing.
(A) Wet lab steps and (B) dry lab steps. The details of each step are described in the text. (Maloyjo Joyraj Bhattacharjee, Basant K. Tiwary, in Biotechnology in Healthcare, 2022)
Data analysis is a crucial component of high-throughput sequencing workflows, as it involves processing, interpreting, and deriving meaningful insights from the vast amounts of sequencing data generated by HTS platforms. The complexity of HTS data, which can range from millions to billions of short DNA or RNA sequences, requires sophisticated bioinformatics tools and computational algorithms to extract biological information accurately and efficiently.
The first step in HTS data analysis involves preprocessing and quality control to ensure the accuracy and reliability of the sequencing data. This includes trimming adapter sequences, filtering out low-quality reads, and removing sequencing artifacts and contaminating sequences. Tools such as FastQC, Trimmomatic, and Cutadapt are commonly used for quality assessment and preprocessing of HTS data (Andrews et al., 2010; Bolger et al., 2014; Martin, 2011).
Once the raw sequencing data has been preprocessed, the next step is read mapping and alignment, where the short sequence reads are aligned to a reference genome or transcriptome to identify their genomic or transcriptomic origins. This process involves aligning each read to the reference sequence, allowing for mismatches, insertions, and deletions. Popular read mapping algorithms include Bowtie, BWA, and HISAT2, which employ different strategies for efficient and accurate alignment of short reads (Langmead et al., 2009; Li and Durbin, 2009; Kim et al., 2015).
Variant calling is a critical step in HTS data analysis, particularly in genomic studies, where it involves identifying single nucleotide variants (SNVs), insertions, deletions, and structural variations in the sequenced genomes. Variant calling algorithms such as GATK, FreeBayes, and VarScan utilize statistical models and machine learning approaches to detect variants from aligned sequencing reads, accounting for sequencing errors, read depth, and mapping quality (McKenna et al., 2010; Garrison and Marth, 2012; Koboldt et al., 2012).
In transcriptomic studies, HTS data analysis involves quantifying gene expression levels and identifying differentially expressed genes between experimental conditions. This typically requires mapping RNA-seq reads to a reference transcriptome and estimating transcript abundances using tools such as Salmon, Kallisto, and RSEM (Patro et al., 2017; Bray et al., 2016; Li et al., 2011). Subsequent differential expression analysis employs statistical methods such as DESeq2, edgeR, and limma to identify genes that are significantly upregulated or downregulated between experimental groups (Love et al., 2014; Robinson et al., 2010; Ritchie et al., 2015).
To gain biological insights from HTS data, functional annotation and pathway analysis are performed to annotate genes, predict their functions, and identify enriched biological pathways or gene ontology terms. Tools such as DAVID, Enrichr, and g:Profiler are commonly used for functional enrichment analysis, allowing researchers to interpret the biological significance of differentially expressed genes or genetic variants (Huang et al., 2009a, 2009b; Chen et al., 2013; Raudvere et al., 2019).
While HTS offers unprecedented opportunities for genomic research, it also presents several challenges, particularly in data analysis. Managing and interpreting large volumes of sequencing data requires sophisticated computational methods and robust bioinformatics infrastructure. Additionally, ensuring data quality and reproducibility is paramount, necessitating rigorous validation and benchmarking of analysis pipelines. However, advancements in machine learning, cloud computing, and data visualization are enhancing the capabilities of bioinformatics tools and enabling researchers to extract deeper insights from HTS data.
Is NGS and HTS the same?
NGS and HTS refer to the same technology: next-generation sequencing and high-throughput sequencing are synonymous terms used interchangeably in the field of genomics.
What is the purpose of high throughput sequencing?
The purpose of High-Throughput Sequencing is to rapidly sequence DNA and RNA molecules on a massive scale, revolutionizing genomic research. This technology enables researchers to efficiently analyze entire genomes or transcriptomes, providing insights into genetic variation, gene expression, and other biological processes.
References:


