
 Sample Submission Guidelines
Sample Submission Guidelines
What is Amplicon Sequencing
Amplicon sequencing involves sequencing specific lengths of PCR products or captured fragments to analyze variations within the sequences. This technique allows for high-coverage sequencing of target regions tailored to different requirements and can detect low-frequency mutations. Currently, amplicon sequencing primarily encompasses functional gene sequencing and targeted sequence-directed capture fragment sequencing.
Amplicon sequencing is based on NGS technology or PacBio SMRT sequencing. The ultra-deep sequencing of amplicons (PCR products) allows efficient variant identification and characterization. This technique has a wide range of applications, including 16S/18S/ITS gene sequencing, SNP genotyping, CRISPR sequencing, somatic/complex variant discovery, antibody screening sequencing, immune repertoire sequencing, et al.
Whether you would like to detect the diversity of microbial communities or discover rare somatic mutations in complex samples. CD Genomics could provide professional, cost-efficient and high-speed amplicon sequencing services to meet your project requirements.
Advantages of Amplicon Sequencing
- Providing high-sensitive detection levels through ultra-deep sequencing
- Achieving high coverage by sequencing hundreds of thousands of amplicons per reaction
- Cost-efficient and fast turnaround time
- Microbial culture is not necessary
- Wide range of amplicons from 100 bp to 10 kb
Amplicon Sequencing Workflow
CD Genomics employs multiple platforms to provide the fast and accurate amplicon sequencing services and bioinformatics analysis. Our highly experienced experts execute quality management, following every procedure to ensure high quality results. The general workflow for amplicon sequencing is outlined below.
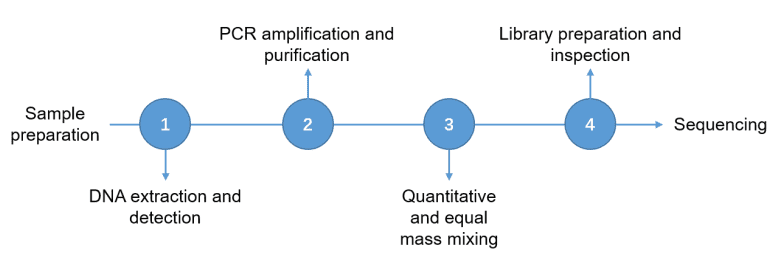
Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Amplicon Size: 100-250 bp:
|
 |
Bioinformatics Analysis We provide customized bioinformatics analysis including: For diversity detection
|
Analysis Pipeline
Amplicon sequencing facilitates various bioinformatic analyses. Depending on your specific needs, we can conduct the necessary data analyses. For instance, the data analysis pipeline for amplicon sequencing can be exemplified by variant identification.
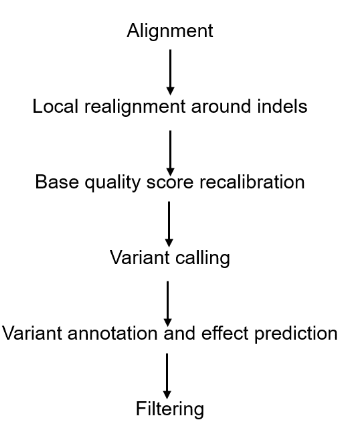
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Amplicon Sequencing for your writing (customization)
References:
- Betge J, Kerr G, Miersch T, et al. 2015. Amplicon sequencing of colorectal cancer: variant calling in frozen and formalin-fixed samples. Plos One 10:e0127146.
- Ison SA, Delannoy S, Bugarel M, et al. 2016. Targeted amplicon sequencing for single-nucleotide-polymorphism genotyping of attaching and effacing Escherichia coli O26:H11 cattle strains via a high-throughput library preparation technique. Appl Environ Microbiol 82:640-649.
Partial results are shown below:

The taxonomy distribution of all sample in Phylum classification level.

Species abundance Heatmap.

Rarefaction curve of the sequenced reads for samples (The above figure) & The depth of the sequencing samples (The below figure).

Boxplot analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

PCoA analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

UPGMA clustering tree.

Mean proportion of treated and control group.

Cladogram.

LDA SCORE.
1. What is the difference between targeted sequencing and amplicon sequencing?
Amplicon sequencing involves the PCR amplification of specific genomic regions followed by sequencing, which ensures high specificity and on-target rates due to the precise design of primers. It is particularly suitable for analyzing small, defined regions of the genome, such as in genetic variation analysis and microbial profiling. In contrast, targeted sequencing encompasses methods like hybrid capture and probe-based enrichment to selectively sequence larger genomic regions or multiple genes without prior amplification. This allows for a more comprehensive analysis of selected areas, but may have variable on-target rates depending on the efficiency of the enrichment process. Amplicon sequencing, by its nature, achieves superior on-target rates in contrast to other targeted sequencing methodologies, attributing this efficiency to the precise design of primers. This approach finds particular applicability in tasks like genotyping via sequencing, as well as the discernment of germline single nucleotide polymorphisms (SNPs), insertions and deletions (indels), and known genetic fusions.
2. What are the primary applications of Amplicon Sequencing?
Amplicon sequencing serves as a pivotal tool in diverse scientific domains, encompassing but not limited to the following applications:
- Genetic Variance Analysis: Unveiling single nucleotide polymorphisms (SNPs), insertions, deletions, and other hereditary genetic modifications.
- Microbiome Investigations: Profiling microbial communities by sequencing marker genes like the 16S ribosomal RNA.
- Oncological Inquiry: Spotting somatic mutations and genetic modifications within tumor specimens.
- Hereditary Disease Research: Exploring the genetic underpinnings of inherited disorders.
- Environmental Surveys: Gauging biodiversity and identifying specific organisms within environmental specimens.
3. What is the difference between Amplicon Sequencing and Whole-Genome Sequencing (WGS)?
Amplicon sequencing targets specific genomic regions by amplifying them with PCR before sequencing, allowing for high specificity and depth in analyzing small, defined regions, such as in detecting mutations or profiling microbial communities. In contrast, whole-genome sequencing (WGS) sequences the entire genome without prior selection or amplification, providing a comprehensive view of all genetic information, which is ideal for discovering novel variants and obtaining a complete genetic profile, but it is more resource-intensive and less focused on specific areas of interest.
4. How do you choose the target regions for Amplicon Sequencing?
The selection of target segments in Amplicon Sequencing hinges on the study's objectives and the biological significance of these segments. Pertinent factors include associations with diseases, genetic markers, regions of notable variability, and functional relevance. Collaborating with bioinformaticians and utilizing databases like dbSNP and ClinVar can facilitate precise target region identification.
5. What types of bioinformatic analyses can be performed with Amplicon Sequencing data?
- Variant identification: Recognizing SNPs, insertions, deletions, and diverse genetic variances.
- Analysis of microbial diversity: Evaluating the constitution and prevalence of microbial consortia.
- Phylogenetic investigation: Researching evolutionary connections among sequences.
- Functional elucidation: Associating genetic variances with plausible functional implications.
6. Can Amplicon Sequencing detect rare variants?
Without a doubt, Amplicon Sequencing displays notable sensitivity, allowing the detection of infrequent variants found at low occurrences. This trait renders it applicable for situations like the spotting of mutations in cancer and the evaluation of microbial diversity.
Full-length 16S rRNA gene sequencing reveals spatiotemporal dynamics of bacterial community in a heavily polluted estuary, China
Journal: Environmental Pollution
Impact factor: 8.071
Published: 15 April 2021
Background
The Liaohe Estuary faces severe inorganic nitrogen and heavy metal pollution, impacting its ecosystem and marine biodiversity. This study uses SMRT sequencing to analyze bacterial community dynamics across seasons and lifestyles, aiming to understand the impact of environmental factors and pollution on the estuarine ecosystem.
Methods
- 60 samples
- 4 L seawater
- DNA extraction
- 16S rRNA full-length gene sequencing
- PacBio Sequel system
- Alpha diversity Analysis
- PCoA and UPGMA
- CCA
- Analysis of similarities (ANOSIM)
Results
After filtering, 449,089 clean reads were generated from sediment and seawater samples, with sediment and seawater yielding 9,803 and 17,950 OTUs respectively. Alpha diversity was higher in sediment samples than seawater samples in both wet and dry seasons. Bacterial communities differed between sediment and seawater, with distinct dominant phyla in each. Seasonal and habitat variations significantly influenced microbial community structures, with lifestyle-based differences observed mainly in the dry season.
Clustering and PCoA analyses revealed significant spatial differences in sediment bacterial communities between the N and O areas, regardless of season, with Gillisia being the core bacterium in the N area.
In seawater, bacterial community relationships were more complex due to space, season, and lifestyle factors, with significant differences observed among free-living (FL) and particle-attached (PA) bacteria, especially in the O area and dry season. Specific bacteria showed seasonal lifestyle transitions, and environmental factors like Cu, Zn, and salinity significantly influenced community structures.
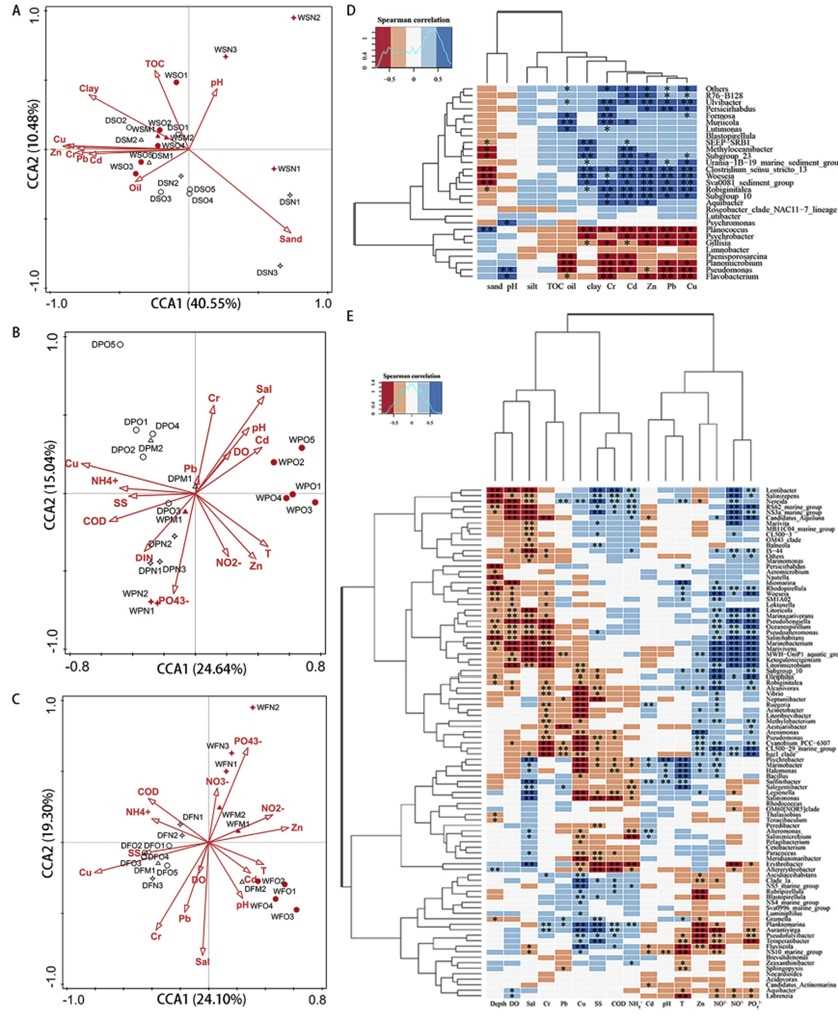 Fig 1. CCA of the relationship between environmental factors and the bacterial community at the species level. Heatmap of Spearman's rank correlation between bacterial genus and chemical characteristics in sediment (D) and seawater (E) samples.
Fig 1. CCA of the relationship between environmental factors and the bacterial community at the species level. Heatmap of Spearman's rank correlation between bacterial genus and chemical characteristics in sediment (D) and seawater (E) samples.
Conclusion
This study reveals distinct bacterial communities in sediment and seawater of the Liaohe Estuary, influenced by habitat, space, season, and lifestyle, with heavy pollution significantly impacting nearshore bacterial diversity and composition.
Reference:
- Hongxia M, Jingfeng F, Jiwen L, et al. Full-length 16S rRNA gene sequencing reveals spatiotemporal dynamics of bacterial community in a heavily polluted estuary, China. Environmental Pollution, 2021, 275: 116567.
Here are some publications that have been successfully published using our services or other related services:
Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Journal: Cell Death & Disease
Year: 2024
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
See more articles published by our clients.


