
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics specializes in NGS-BSP services, offering advanced capabilities for comprehensive DNA methylation analysis. Our service utilizes state-of-the-art NGS technology to accurately map and quantify methylated cytosines, providing detailed insights into epigenetic modifications across various biological samples. This enables precise characterization of gene regulation mechanisms and facilitates research into complex diseases.
The Introduction of NGS-BSP
DNA methylation strongly affects chromatin structure and the regulation of gene expression. For many years, Bisulfite Sequencing PCR (BSP) has served as the "gold standard" for measuring DNA methylation of specific regions. BSP has high reliability and accuracy, and can accurately determine the methylation status of each CpG site in the fragment. The Next Generation Sequencing-based Bisulfite Sequencing PCR (NGS-BSP) can find more precise methylation modification information, and is applicable to some genes that are known methylated detection regions, and is also suitable for exploring significant methylation regions of genes.
Experimental Principle of BSP:
Genomic DNA is treated with bisulfite, converting all unmethylated cytosines to uracil while methylated cytosines remain unchanged. Subsequently, primers are designed at both ends of CpG islands for PCR amplification. The purified target products are then cloned using TA cloning. Each clone is then selected for sequencing to identify positive clones. Finally, the obtained sequences are compared with the original sequence to determine the methylation sites and quantify the degree of methylation.
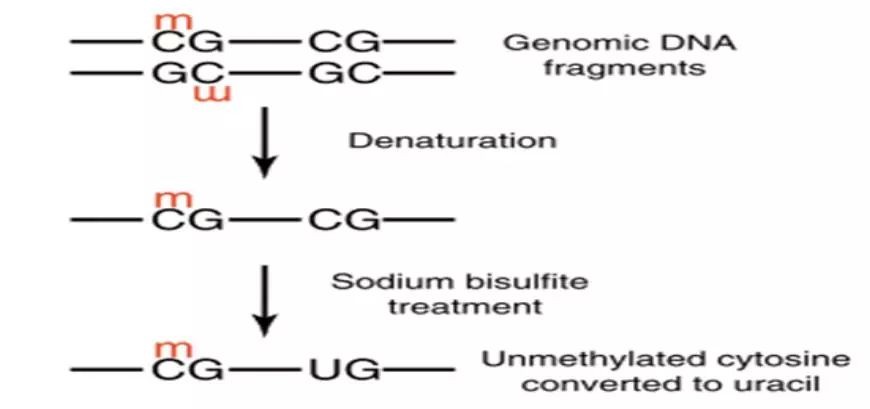 Figure 1. Experimental Principle of BSP.
Figure 1. Experimental Principle of BSP.
Advantages of Our NGS-BSP Service
- Quickly detect all methylation sites
- High throughput, High accuracy, Simple and intuitive results
- More cost-effective and time-saving than original BSP if testing a lot of samples
Applications of NGS-BSP
- Epigenetics Research
- Clinical Applications
- Environmental and Exposome Studies
- Agricultural and Livestock Research
NGS-BSP Workflow
In brief, BSP primers were designed using the online MethPrimer software. Genomic DNA (1 ug) was converted using the ZYMO EZ DNA Methylation-Gold Kit (ZYMO) and one twentieth of the elution products is used as templates for PCR amplification. For each sample, BSP products of multiple genes were generated, pooled equally and subjected to adaptor ligation. Barcoded libraries from all samples were sequenced on the Illumina Hiseq platform using paired-end 150 bp strategy.

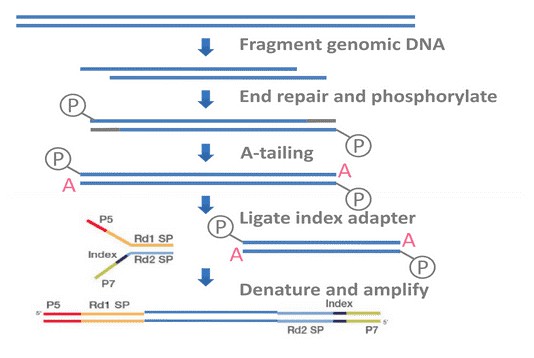
Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
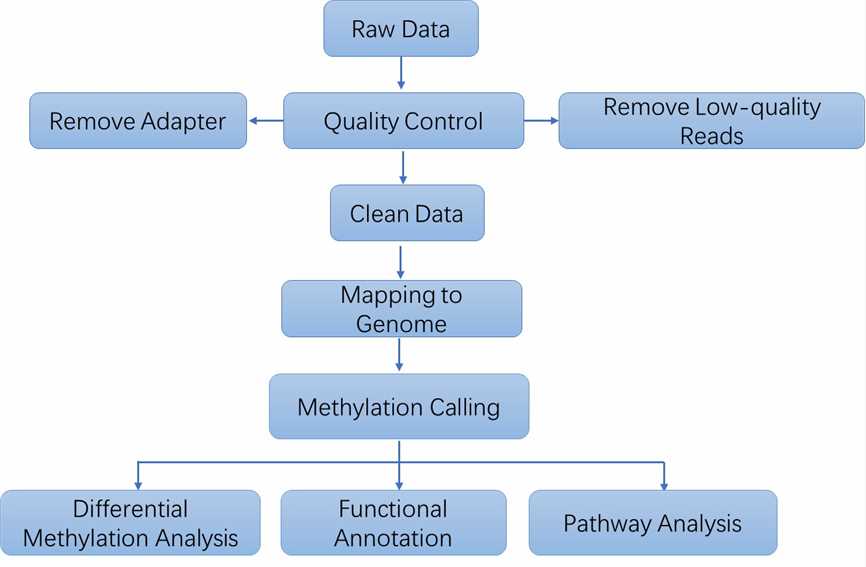
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in NGS-BSP for your writing (customization)
CD Genomics offers consultation with you at every stage of the process to ensure that you make the most use of your data. We offer professional assistance from planning experiments to sequencing and further data analysis. If you have any requirements do let us know and we will be there to assist you. We'd love the opportunity to work with you. Please contact us for more information and a detailed quote.
References
- Pan X, Gong D, Nguyen DN, et al. Early microbial colonization affects DNA methylation of genes related to intestinal immunity and metabolism in preterm pigs. DNA Research, 2018.
- Zhang S, Shen L, Xia Y, et al. DNA methylation landscape of fat deposits and fatty acid composition in obese and lean pigs. Scientific Reports, 2016, 6:35063.
Partial results are shown below:
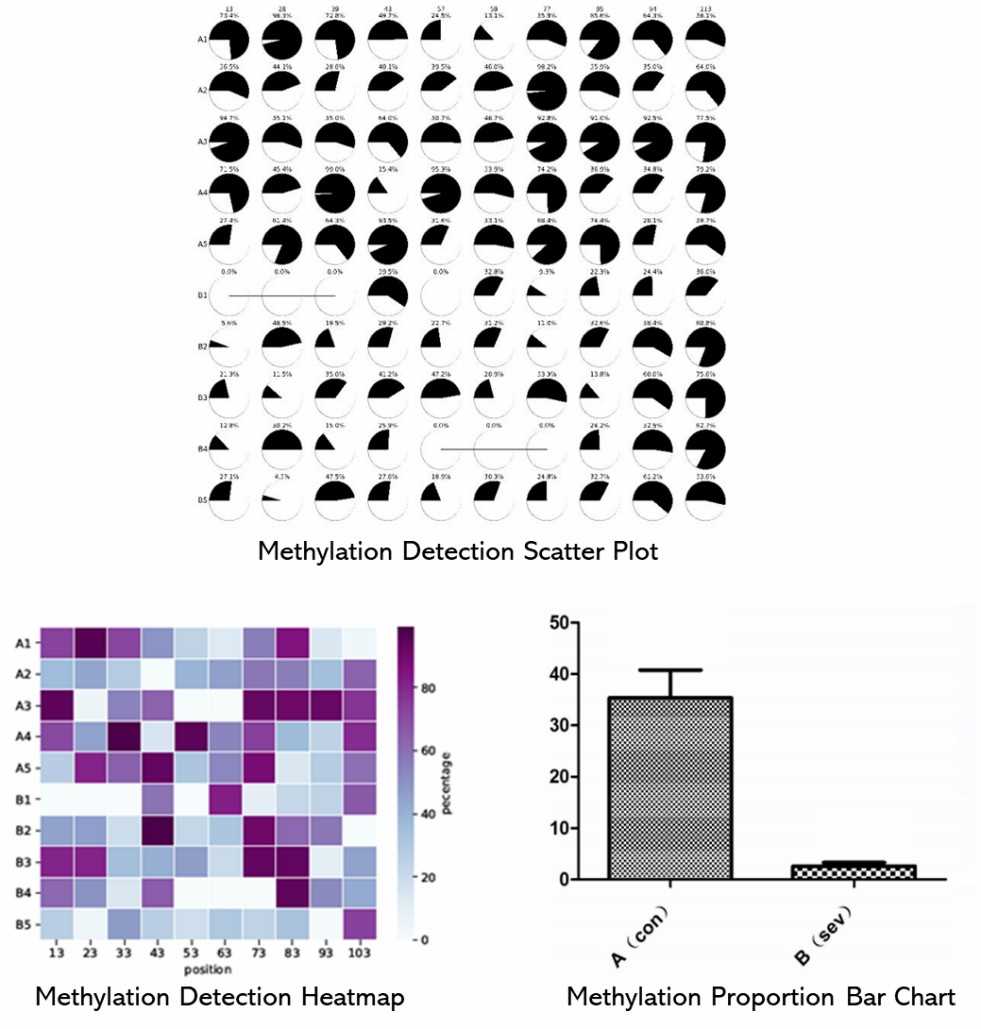
1. How is the bisulfite treatment process carried out?
Bisulfite treatment involves the exposure of DNA to sodium bisulfite, leading to the deamination of unmethylated cytosine residues to uracil, while leaving methylated cytosines unaltered. This chemical modification enables the differentiation of methylated and unmethylated cytosines during sequencing, facilitating the subsequent analysis of DNA methylation patterns.
2. What are the commonly used tools for the analysis of NGS-BSP data?
Various specialized tools and software are utilized for the analysis of NGS-BSP data, including:
- FastQC: for assessing the quality of raw sequencing data.
- Trim Galore: for removing low-quality reads and adapter sequences.
- Bismark: for aligning bisulfite-treated reads to the reference genome and determining DNA methylation levels.
- SAMtools: for managing alignment files.
- MethylKit: for conducting differential methylation analysis and visualizing results.
- IGV (Integrative Genomics Viewer): for visualizing methylation patterns.
- HOMER: for annotating functional genomic elements.
- KEGG and GO Analysis: for elucidating biological pathways influenced by DNA methylation changes.
3. What are the key considerations for DNA extraction in NGS-BSP?
For the successful implementation of NGS-BSP, the extraction of high-quality DNA stands as a cornerstone. It is imperative to factor in the following considerations:
Employing DNA extraction techniques that deliver DNA of elevated purity and integrity. Preventing contamination with RNA, proteins, or other extraneous impurities. Ensuring the DNA is present in adequate quantity and concentration for subsequent applications.
4. How is the efficiency of bisulfite conversion assessed?
The efficacy of bisulfite conversion can be evaluated through various means:
- Incorporating unmethylated control DNA and ensuring complete conversion (absence of residual cytosines).
- Sequencing bisulfite-converted DNA and determining the conversion rate (percentage of unmethylated cytosines converted to uracil).
- Employing spike-in controls or internal standards to monitor the efficiency of conversion.
5. How do you design primers for NGS-BSP?
Designing primers for NGS-BSP demands special attention owing to the sequence modifications induced by bisulfite treatment:
- Devising primers devoid of cytosines to accommodate their conversion to uracils (which switch to thymines post-PCR).
- Leveraging software tools like MethPrimer or BiSearch for the creation of bisulfite-specific primers.
- Verifying primer efficiency and specificity through validation with bisulfite-treated DNA.
Promoter methylation changes in the placenta involved in the relationship between prenatal depression and small for gestational age
Journal: BMC Pregnancy and Childbirth
Impact factor: 3.105
Published: 02 October 2022
Background
Depression and anxiety during pregnancy affect 10% to 40% of pregnant women and are linked to low birth weight and SGA. Increased glucocorticoid levels due to HPA axis dysfunction may impact fetal development via the placenta, which regulates glucocorticoid exposure through HSD11β2. Thyroid hormones (HPT axis) and melatonin (HPA axis) also play roles. Maternal emotions may cause placental epigenetic changes. This study will utilize next-generation sequencing and DNA methylation analysis techniques to examine the relationship between SGA and maternal emotions, specifically investigating methylation changes in genes such as CRH, HSD11β2, SLC16A10, DIO3, and MTNR1B.
Materials & Methods
Sample Preparation
- Pregnant women
- Placenta sample collection
- DNA extraction
Sequencing
- BSP primer design
- NGS-BSP
- Illumina HiSeq
- Shapiro–Wilk test
- Differential analysis of methylation levels
- Statistical analysis
Results
A nested case-control study found that maternal depression in the second trimester is a risk factor for SGA. Placentas were divided into four groups based on depression and SGA status, with 17 samples each. Methylation levels of CRH and DIO3 genes were higher in depressed mothers, while HSD11β2 levels were higher in non-depressed mothers. Methylation of CRH and HSD11β2 correlated with depression, and DIO3 methylation correlated with SGA, suggesting maternal depression may influence fetal development through these epigenetic changes.
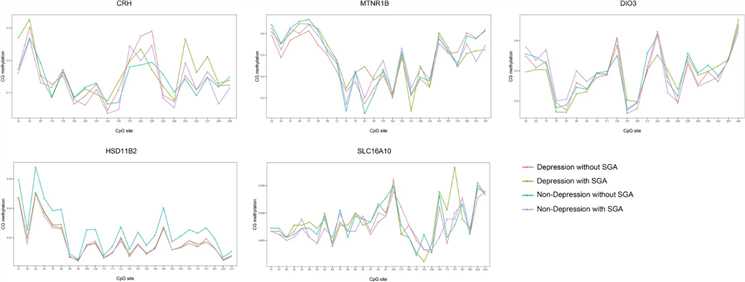 Figure 1. Methylation of CRH, HSD11β2, SLC16A10, DIO3, and MTNR1B genes in the placenta.
Figure 1. Methylation of CRH, HSD11β2, SLC16A10, DIO3, and MTNR1B genes in the placenta.
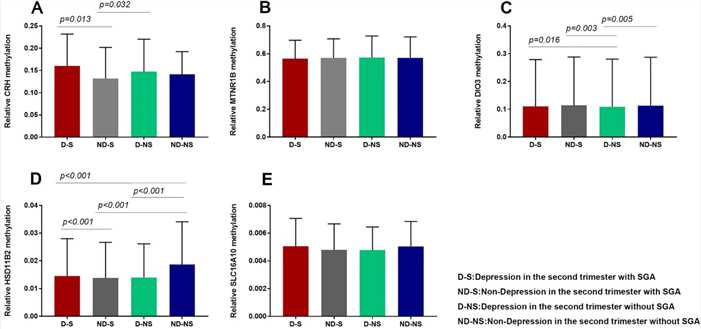 Figure 2. Comparison of placental gene (CRH, HSD11β2, DIO3, SLC16A10, and MTNR1B) methylation levels in four groups.
Figure 2. Comparison of placental gene (CRH, HSD11β2, DIO3, SLC16A10, and MTNR1B) methylation levels in four groups.
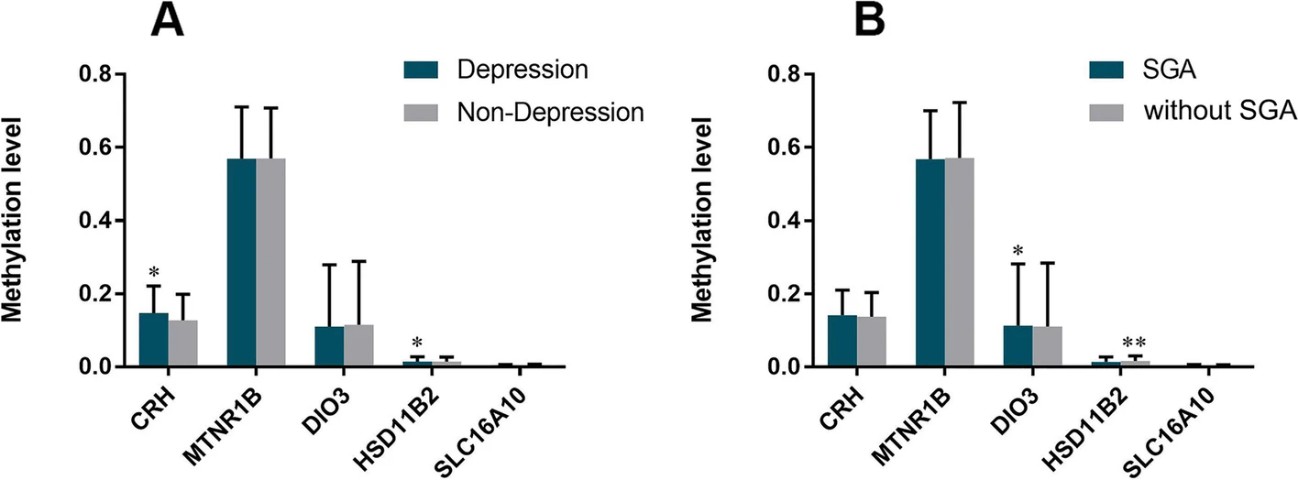 Figure 3. Comparison methylation level of CRH, HSD11β2, DIO3, SLC16A10, MTNR1B according to depression of mother and SGA birth.
Figure 3. Comparison methylation level of CRH, HSD11β2, DIO3, SLC16A10, MTNR1B according to depression of mother and SGA birth.
Conclusion
Maternal depression in the second trimester disrupts the HPA and HPT axes, increasing SGA risk through placental gene methylation changes (DIO3, HSD11β2, CRH). Early screening and intervention are crucial due to potential long-term effects on offspring behavior and development. Healthcare professionals should prioritize emotional support for pregnant women and monitor offspring of depressed mothers for developmental impacts. Increasing sample sizes in future research will improve the validity of these findings.
Reference
- Yang J, Xu A, Zhang Y, et al. Promoter methylation changes in the placenta involved in the relationship between prenatal depression and small for gestational age. BMC Pregnancy and Childbirth. 2022, 22(1):741.
Here are some publications that have been successfully published using our services or other related services:
Variable Response of Norepinephrine Transporter to Traumatic Stress and Relationship to Hyperarousal
Journal: Frontiers in Behavioral Neuroscience
Year: 2021
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


