
 Sample Submission Guidelines
Sample Submission Guidelines
Microbial Whole Genome Sequencing
With decades of experience in the fields of genome sequencing, CD Genomics is devoted to providing the accurate and affordable microbial whole genome sequencing service. We combine both Illumina (short reads) and PacBio (long reads) platforms for microbial re-sequencing and complete genome de novo sequencing. Our strong expertise is enhanced by flexible sequencing strategies and professional bioinformatics pipelines.
The Introduction of Microbial Whole Genome Sequencing
There are great differences between microorganisms and higher eukaryotes. Besides their smaller genome, most bacteria have a single circular chromosome (sometimes more than one chromosomes, linear chromosomes, or combinations of linear and circular chromosomes). The genes in microbial genomes are usually a single continuous stretch of DNA, although several types of introns seldom exist in bacterial genome. The presence of plasmids in bacterial genome is another major difference. Plasmids, the circular extra-chromosomal DNA, can be transferred via horizontal DNA transfer, mediating the rapid evolution of microorganisms. Virus is a non-cellular infectious organism consisting of a core of DNA or RNA surrounded by a protein coat.
Microbial whole genome sequencing yields tons of data enabling a comprehensive evaluation of all genetic features of an isolated microorganism. Shotgun sequencing strategy is a primary method of microbial whole genome sequencing. The sequencing steps do not need labor-intensive mapping and cloning, which saves tremendous time and money. Furthermore, high-throughput sequencing allows us to sequence hundreds of bacteria or viruses at the same time with the power of multiplexing. In whole genome shotgun sequencing, the whole genome is broken up into small fragments for sequencing, and then assembled together by computational methods based on the overlapped regions, hence not requiring a reference genome. PacBio SMRT technology enables us to provide bacterial de novo whole genome sequencing and fungal de novo whole genome sequencing that generate more accurate and contiguous sequences.
Microbial whole genome sequencing is crucial for precise microbial identification, the generation of complete reference genomes (de novo sequencing), comparative genomic studies (re-sequencing), and genomic exploitation. Comparative genomic studies can identify individual genetic variations and large-scale structural variations within a population for which a reference genome is available. Evolutionary characteristics and phylogenetic relationships can be hence inferred. Microbial whole genome sequencing provides the possibility of gene finding and annotation. After multiple genes are explained, novel biochemical pathways that may be beneficial for medicine and biotechnology will likely be identified.
Advantages of Microbial Whole Genome Sequencing
- A time-effective and cost-efficient approach
- Broad applications: de novo sequencing, gene annotations, comparative genomic studies, evolutionary studies of microorganisms, etc.
- Drug discovery and development: assess the contribution of DNA on pathogenesis, and understand the role of mobile elements in drug resistance and transmission
Application of Microbial Whole Genome Sequencing
- Pathogen Identification: Swift and precise determination of pathogen identity through comparison with established genome databases, facilitating clinical diagnoses and epidemiological inquiries.
- Antibiotic Resistance Monitoring: Identification and analysis of resistance genes to unveil microbial resistance mechanisms, furnishing guidance for clinical management and surveillance of resistance dissemination.
- Genome Comparisons: Comparative analysis of genomes across different strains to delineate genetic variations, aiding comprehension of microbial evolution, genetic diversity, and population dynamics.
- Functional Genomics: Discovery of functional genes and metabolic pathways within genomes to decipher microbial biological traits and ecological roles.
- Environmental Pollution Monitoring: Sequencing analysis of environmental microbial communities to monitor the effects of pollutants and microbial responses, evaluating environmental integrity.
- Drug Discovery: Harnessing microbial genome data for the exploration of novel drug targets and natural products, expediting drug development and discovery endeavors.
- Biosecurity and Control: Vigilant monitoring and preemptive detection of potential bioterrorism or biosecurity threats, ensuring public and national security.
- Food Safety Monitoring: Surveillance and detection of microbial contamination in food to uphold food safety standards and safeguard consumer health.
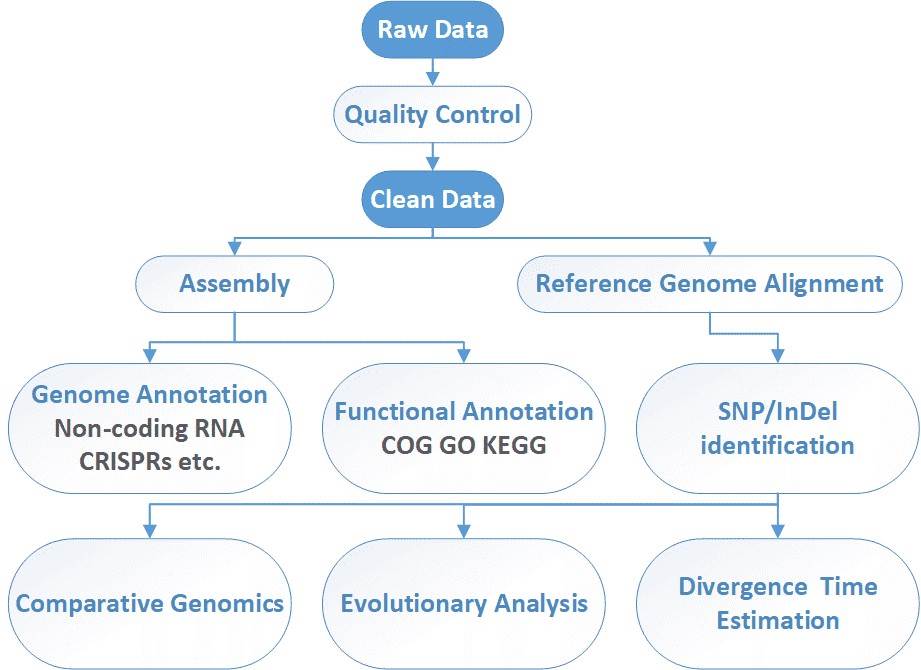
Microbial Whole Genome Sequencing Workflow
CD Genomics can offer integrated genome sequencing services for bacteria, yeast, fungi, phage and virus. Our highly experienced expert team executes high quality management following every procedure to ensure confident and unbiased results with Illumina HiSeq and/or PacBio SMRT system. The general workflow for microbial whole genome sequencing is outlined below.

Service Specification
Sample Requirements
|
|
 |
Sequencing
|
 |
Data Analysis
We provide customized bioinformatics analysis:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Microbial Whole Genome Sequencing for your writing (customization)
Reference:
- Fraser C. M., et al. Microbial genome sequencing. Nature, 2000, 406(6797): 799.
Demo Results
Partial results are shown below:

Distribution of base quality.

Distribution of base content.

Shared SNP number between samples.

SNP mutation type distribution.
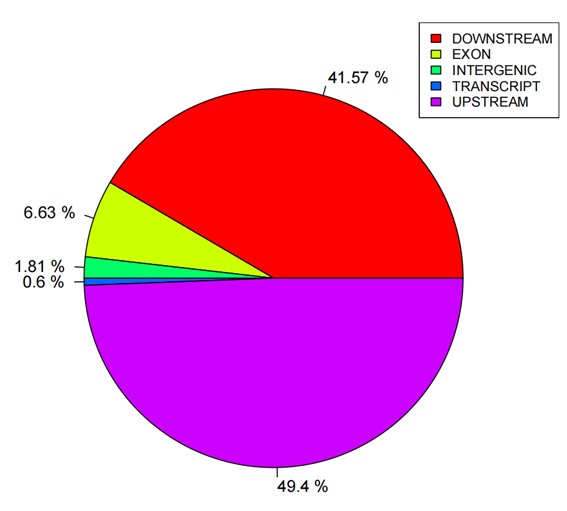
Statistics pie of SNP annotations.

Shared InDel number between samples.

InDel length distribution in both the whole genome scale and CDS regions.

Statistics pie of InDel annotations.
Microbial Whole Genome Seq FAQs
1. How to prepare samples?
You can submit pure cultured microorganisms or extracted genomic DNA samples for microbial whole genome sequencing. For cultured microorganisms, the purified cells should be spun down in a 1.5 mL tube not containing media. We will need a minimum of 1 million cells, and the more cells you can offer, the better. For genomic DNA samples, the recommended amount for submission is 2 µg or more with a concentration of more than 20 ng/µl. The ratio of OD260/OD280 is between 1.8 and 2.0. Samples should be shipped frozen on dry ice.
Faeces samples: store the faeces samples below -80℃. Empirically, fresh faeces contribute to isolating preferable DNA.
2. How to increase the accuracy of genome assembly when utilizing PacBio SMRT system?
The raw read error rate of PacBio SMRT system is substantially higher at around 14% compared with the 0.1 to 1% error rate of other leading sequencing systems. However, the error model is stochastic, so very high quality reads across all bases can be achieved in the consensus sequence. Additionally, the SMRT sequencing system is capable of sequencing regions of high GC content, leading to much more uniform coverage of the genome.
There are three ways to ensure the accuracy of genome assembly: (i) prior to assembly, correct sequences in the consensus sequence; (ii) correct the results of sequence assembly utilizing sequencing data; (iii) correct the results of sequence assembly utilizing high quality next generation sequencing data. After the three corrections, the accuracy of final sequence assembly can reach 99.99%.
3. How to achieve zero gap?
Currently, the complete sequence map of more than 90% bacterial strains can be constructed by making use of a combination of Illumina HiSeq and PacBio SMRT systems. The complete sequence map of the rest 10% bacterial strains can be achieved with Sanger sequencing data.
4. Can complete genome assembly be achieved even in the regions of high or low GC content, as well as repetitive sequences?
Pacbio RS II system can overcome the foregoing challenges. CD Genomics has completed hundreds of bacterial genome assembly cases without gap.
5. What are the advantages of microbial whole genome sequencing in clinical practice?
Whole genome sequencing has been a routine tool for clinical microbiology. It can reduce diagnostic time and improve control and treatment. It describes and improves our understanding of microbial evolution, outbreaks and transmission events. The conventional procedures for whole genome sequencing often include multiple cultivation and incubation steps followed by species identification, susceptibility testing and typing, which may take several weeks. A number of other approaches (such as PCR based methods) are cheap and rapid, but limited in the sensitivity. Although whole genome sequencing is still too expensive, the price and turnaround time will most likely fall in terms of the competition among sequencing platforms.
Reference:
- Hasman H., et al. Rapid whole genome sequencing for the detection and characterization of microorganisms directly from clinical samples. Journal of clinical microbiology, 2013: JCM. 02452-13.
Microbial Whole Genome Seq Case Studies
The Genome Sequence of the Highly Acetic Acid-Tolerant Zygosaccharomyces bailii-Derived Interspecies Hybrid Strain ISA1307, Isolated from a Sparkling Wine Plant
Journal: DNA Research
Impact factor: 5.404
Published: June 2014
Background
This work described the genome sequencing and annotation of the yeast strain ISA1307, isolated from a sparkling wine production plant. This strain, formerly considered as the Zygosaccharomyces bailii species, is an interspecies hybrid between Z. bailii and a closely related species.
Methods
- The prototrophic yeast isolates ISA1307
- Z. bailii ATCC58445T
- S. cerevisiae BY4714 and BY4743
- Quantification of genomic DNA
- Pulsed field gel electrophoresis (PFGE)
- Whole genome shotgun sequencing
- Illumina
- Karyotyping of the ISA1307 strain
- Genome assembly and annotations
Results
1. The ISA1307 Hybrid strain
The comparison of sequences of the house-keeping genes (including RPB1,RPB2, EF1-α, and β-tublin) revealed that ISA1307 strain is an interspecies hybrid between Z. bailii and a closely related species.
2. Genome assembly
The quantification by flow cytometry revealed that the estimated size of ISA1307 is around 22.0 Mb with S. cerevisiae BY4741 and BY4743 used as a calibration curve (Figure 1A). PFGE profiling of the ISA1307 was performed, and 13 chromosomal bands were observed, with sizes ranging from 733 to 2120 Mb (Figure 1B).
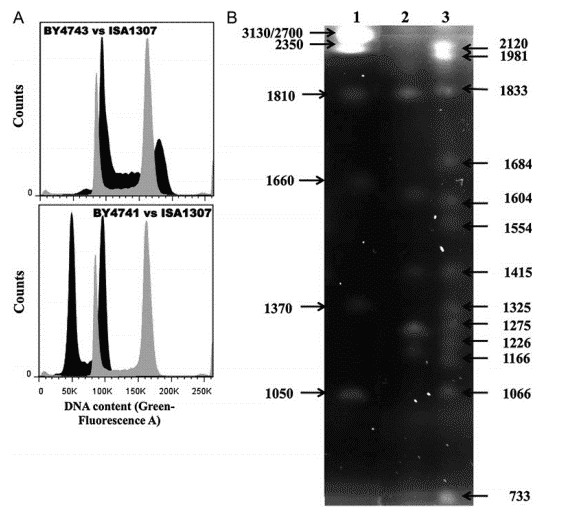 Figure 1. Estimation of genome size and karyotyping of the ISA1307 strain. (A) Representative cell analysis histogram. (B) Karyotype of the reference strain Z. bailii ATCC58445 (lane 2) and of the ISA1307 strain (lane 1).
Figure 1. Estimation of genome size and karyotyping of the ISA1307 strain. (A) Representative cell analysis histogram. (B) Karyotype of the reference strain Z. bailii ATCC58445 (lane 2) and of the ISA1307 strain (lane 1).
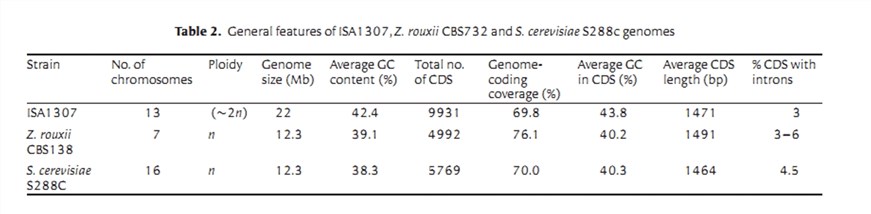
The final reconstructed genome of the ISA1307 strain is distributed over 154 scaffolds. The sum of all scaffolds size is 21241152 bp, which corresponds to 96% of the genome size that was estimated by flow cytometry (Table 1).

3. Annotation
A total of 9,925 genes are predicted to be encoded by the ISA1307 strain, including 4,385 duplicated gene and 1,155 single-copy genes. The predicted functions involve "metabolism and generation of energy", "protein folding, modification and targeting", and "biogenesis of cellular components". The authors further studied the ISA1307 genes and proteins involved in the above functions.
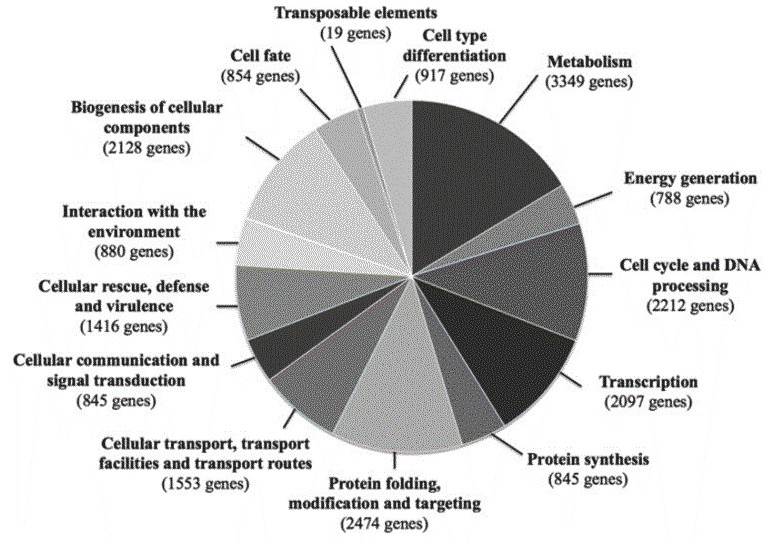 Figure 2. Functional classes of genes predicted to be encoded by the genome of the ISA1307 strain.
Figure 2. Functional classes of genes predicted to be encoded by the genome of the ISA1307 strain.
Conclusion
Accordingly, a cadre of specific reference alleles has been established, utilizing third-generation sequencing technology, which offers a longitudinal thorough exploration of gene loci stretching across numerous thousand base pairs. This novel technique complements second-generation or short-read sequencing techniques, proving to be critically instrumental in deciphering novel, rare, and null alleles.
Reference:
- Mira N. P., et al. The genome sequence of the highly acetic acid-tolerant Zygosaccharomyces bailii-derived interspecies hybrid strain ISA1307, isolated from a sparkling wine plant. DNA Research, 2014, 21(3): 299-313.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Association of the prophage BTP1 and the prophage encoded gene, bstA, with anti-virulence of Salmonella Typhimurium ST313
Journal: Plant Disease
Year: 2020
Broad-Spectrum, Potent, and Durable Ceria Nanoparticles Inactivate RNA Virus Infectivity by Targeting Virion Surfaces and Disrupting Virus–Receptor Interactions
Journal: Molecules
Year: 2023
Over-Expression of UV-Damage DNA Repair Genes and Ribonucleic Acid Persistence Contribute to the Resilience of Dried Biofilms of the Desert Cyanobacetrium Chroococcidiopsis Exposed to Mars-Like UV Flux and Long-Term Desiccation
Journal: Front. Microbiol.
Year: 2019
Putrescine biosynthesis and export genes are essential for normal growth of avian pathogenic Escherichia coli
Journal: BMC Microbiology
Year: 2018
Phenotypic and Draft Genome Sequence Analyses of a Paenibacillus sp. Isolated from the Gastrointestinal Tract of a North American Gray Wolf (Canis lupus)
Journal: Applied Microbiology
Year: 2023
Classification of genera of Pasteurellaceae using conserved predicted protein sequences
Journal: International Journal of Systematic and Evolutionary Microbiology
Year: 2018
See more articles published by our clients.


