
 Sample Submission Guidelines
Sample Submission Guidelines
Besides eukaryotic RNA sequencing service, CD Genomics is also dedicated to providing prokaryotic RNA sequencing service to advance your bacterial gene expression profiling needs by utilizing the latest techniques.
The Introduction of Bacterial RNA Sequencing
In recent years, high throughput sequencing of cDNA libraries (RNA-Seq) has emerged as a powerful technology for profiling gene expression, discovering previously unannotated genes, and mapping transcriptome architecture in a wide variety of bacterial species. With the application of RNA-seq derivative approaches, we can gain biological insights into the bacterial world and aspire to uncover the mysteries involving gene expression, organization and other functional genomic features. Bacterial transcriptome and metatranscriptome information are important for predicting resistance to specific antibiotics, understanding host-pathogen immune interactions, quantifying gene expression changes, and tracking disease progression.
There are obvious differences in cDNA library construction between bacterial RNA-Seq and Eukaryotic RNA-Seq. Bacterial RNA-Seq workflow's first step is selection of mRNA transcripts. Ribo-Zero ribosomal RNA reduction chemistry is used in place a poly-A tail selection which makes this method particularly suited for bacteria mRNA lack a poly-A tail. Following purification, the mRNA is fragmented into small pieces, and copied into first strand cDNA using reverse transcriptase and random primers. Strand specificity is achieved by replacing dTTP with dUTP in the Second Strand Marking Mix (SMM), followed by second strand cDNA synthesis. Through the use of strand-specific RNA-Seq, a more complete understanding of the transcriptome could be achieved, this has the potential to identify new levels of regulation of gene expression.
Advantages of Bacterial RNA Sequencing
- Accurate Gene Structure Annotation: Transcriptome sequencing in prokaryotes can identify small peptides that may be overlooked in genome annotation, providing more precise gene structure information.
- Detection and Annotation of mRNA Non-coding Regulatory Regions: Transcriptome sequencing reveals important regulatory elements in prokaryotic mRNA, such as riboswitches and small RNA binding sites, enhancing our understanding of mRNA regulatory mechanisms.
- Study of Operon Structure and Function: Transcriptome analysis can determine operon structures, advancing our understanding of multifunctional regulation and evolution of polycistronic transcripts.
- Research on Non-coding RNA Regulation: Through prokaryotic sRNA sequencing, we gain insights into the function and mechanisms of these regulatory elements in physiological control.
- Plasticity of RNA Element Functions: Transcriptome sequencing reveals the functional versatility of RNA elements under different conditions, aiding in understanding their roles in diverse environments.
Application of Bacterial RNA Sequencing
- Investigating the molecular regulatory mechanisms of microorganisms related to different stages, phenotypes, and stress responses.
- Improving microbial transcriptome information, including transcript structure determination, prediction of novel transcripts, and identification of non-coding RNAs.
- Gene expression level
- Gene structure level
- Gene function
- Interaction network
- Identification of Differential Genes
- Detection of Non-coding RNAs, etc
Bacterial RNA Sequencing Workflow
The primary stages involved in Bacterial RNA Sequencing commence with the extraction of total RNA from bacterial cells. This operation is succeeded by the elimination of rRNA, which predominates the total RNA, to selectively augment the mRNA population. Subsequently, the mRNA is converted into complementary DNA (cDNA) through reverse transcription. The resultant cDNA is fragmented, adapters are ligated, and PCR amplification is executed to formulate the sequencing library. High-throughput sequencing is subsequently conducted utilizing platforms such as Illumina or PacBio. The concluding phase encompasses a meticulous data analysis process, encompassing quality assurance, sequence alignment, quantification, and differential expression scrutiny.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis Gene structure level analysis
|
Analysis Pipeline
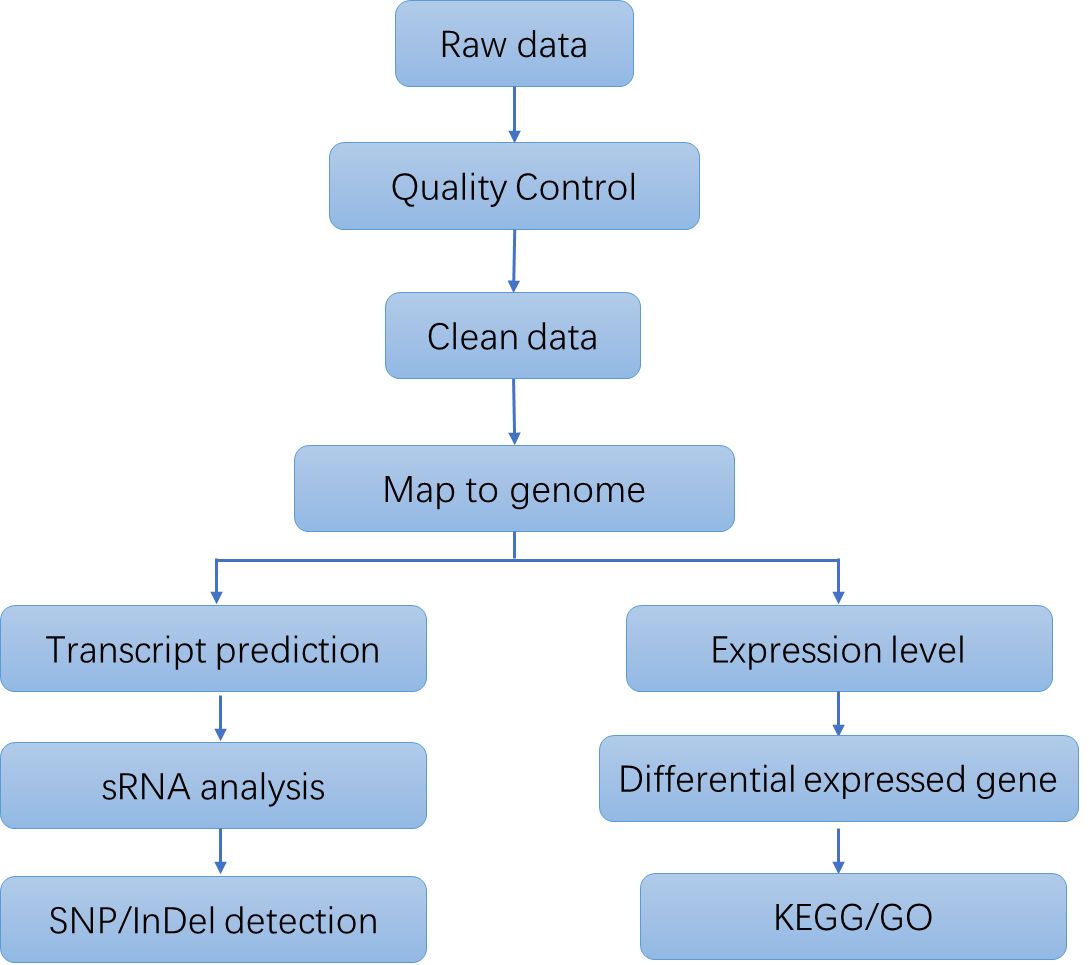
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Bacterial RNA Sequencing for your writing (customization)
CD Genomics provides a fast, one-stop bacterial RNA sequencing solution from the quality control of sample to comprehensive data analysis. Please contact us for more information and a detailed quote.
References
- Kröger C, MacKenzie K D, Alshabib E Y, et al. The primary transcriptome, small RNAs and regulation of antimicrobial resistance in Acinetobacter baumannii ATCC 17978. Nucleic acids research, 2018, 46(18): 9684-9698.
- Liu X, Shen B, Du P, et al. Transcriptomic analysis of the response of Pseudomonas fluorescens to epigallocatechin gallate by RNA-seq. PloS one, 2017, 12(5): e0177938.
Partial results are shown below:

Sequencing quality distribution

A/T/G/C Distribution

IGV Browser Interface

Correlation Analysis Between Samples

PCA Score Plot

Venn Diagram

Volcano Plot

Statistics Results of GO Annotation

KEGG Classification
1. Why is rRNA removal necessary?
The meticulous removal of ribosomal RNA (rRNA) holds pivotal importance in Bacterial RNA Sequencing, owing to its predominant presence in bacterial RNA samples, often exceeding 90% of the total RNA content. As messenger RNA (mRNA)-the primary target of sequencing endeavors-constitutes only a minor fraction of the total RNA pool, effective rRNA depletion is indispensable to mitigate the overwhelming abundance of rRNA sequences in sequencing datasets. Neglecting to eliminate rRNA can significantly jeopardize sequencing precision and compromise the sensitivity of mRNA detection.
2. Is it possible to perform prokaryotic transcriptome sequencing without a reference genome?
No, as it is not practically feasible. Prokaryotic mRNA is commonly polycistronic, making direct assembly unreliable.
3. How does single-cell RNA sequencing differ from Bacterial RNA Sequencing?
Single-cell RNA sequencing stands out from Bacterial RNA Sequencing by its emphasis on studying gene expression at the level of individual cells, exposing cell-to-cell variations. Unlike Bacterial RNA Sequencing, which examines the average gene expression of a bacterial population, the uniqueness of single-cell RNA sequencing lies in its distinct approaches to sample preparation, data analysis, and potential applications.
The claim of primacy of human gut Bacteroides ovatus in dietary cellobiose degradation
Journal: Gut Microbes
Impact factor: 11.724
Published: 22 Jun 2023
Background
Cellulose, a primary constituent of plant cell walls, cannot be digested by mammals without the assistance of cellulolytic bacteria. These bacteria, residing in the gastrointestinal tracts of various animals, including humans, play an essential role in the degradation of cellulose, thereby providing energy to their hosts. Although a substantial number of cellulolytic bacteria have been identified, many remain insufficiently characterized. The key genes responsible for cellulase production are predominantly found within the glycoside hydrolase (GH) families. In humans, cellulase activity has been notably observed in Ruminococcus champanellensis, but further investigation is imperative to fully elucidate its functional mechanisms. Recent advancements, utilizing RNA sequencing (RNA-seq) and 16S rRNA sequencing, are focused on discovering new cellulases and comprehending the intricate processes underlying cellulose degradation in the human gut.
Methods
Sample Preparation:
Lactobacillus rhamnosus GG
Lactobacillus reuteri (LR)
Bifidobacterium longum (BL)
Sequencing:
Microbial analysis
Statistical analysis
Results
To investigate the PULs responsible for cellobiose degradation, BO co-cultured with cellobiose was subjected to RNA sequencing. Results revealed 91 genes with altered expression in response to cellobiose, 19 of which were significantly up-regulated compared to BO grown on glucose. These up-regulated genes were organized into two PULs, PUL1 and PUL2, confirmed by RT-qPCR. PUL1, comprising 15 genes including glycosidase hydrolases (GH5 and GH2) and six hypothetical proteins, and PUL2, comprising five genes including GH16 and GH3, are implicated in cellobiose degradation. Notably, PUL1 had not been previously reported.
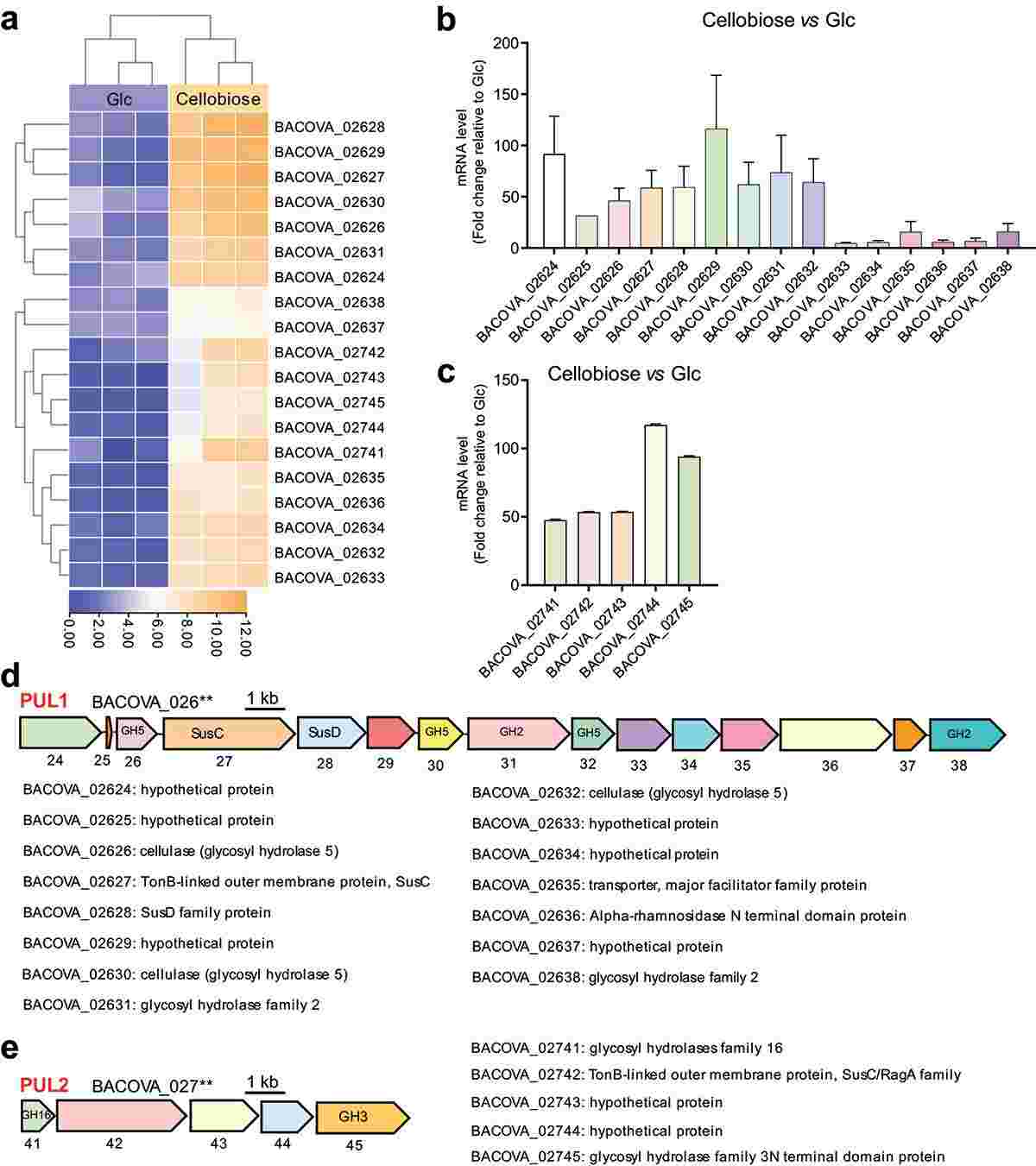 Figure 1. Polysaccharide utilization loci (PULs) were screened by RNA-seq and confirmed by RT-qPCR.
Figure 1. Polysaccharide utilization loci (PULs) were screened by RNA-seq and confirmed by RT-qPCR.
To elucidate the specific pathways by which cellobiose degradation is facilitated in the bacterium, recombinant proteins representing each polysaccharide utilization locus (PUL) were isolated and immune sera produced for characterization. Evidently, the proteins BACOVA_02626GH5, BACOVA_02630GH5, and BACOVA_02745GH3 exhibited specific upregulation in the presence of cellobiose, implicating their direct involvement in the degradation of this substrate. Through a combination of immunofluorescence and Western blot analyses, it was established that BACOVA_02630GH5 localizes predominantly to the cell membrane, while BACOVA_02626GH5 and BACOVA_02745GH3 were expressed at relatively lower levels within the cell. Enzymatic assessments further confirmed the ability of BACOVA_02626GH5, BACOVA_02630GH5, and BACOVA_02745GH3 to catalyze the conversion of cellobiose into glucose. These collective observations underscore the indispensable role played by these proteins in the cellobiose degradation pathway within the bacterium.
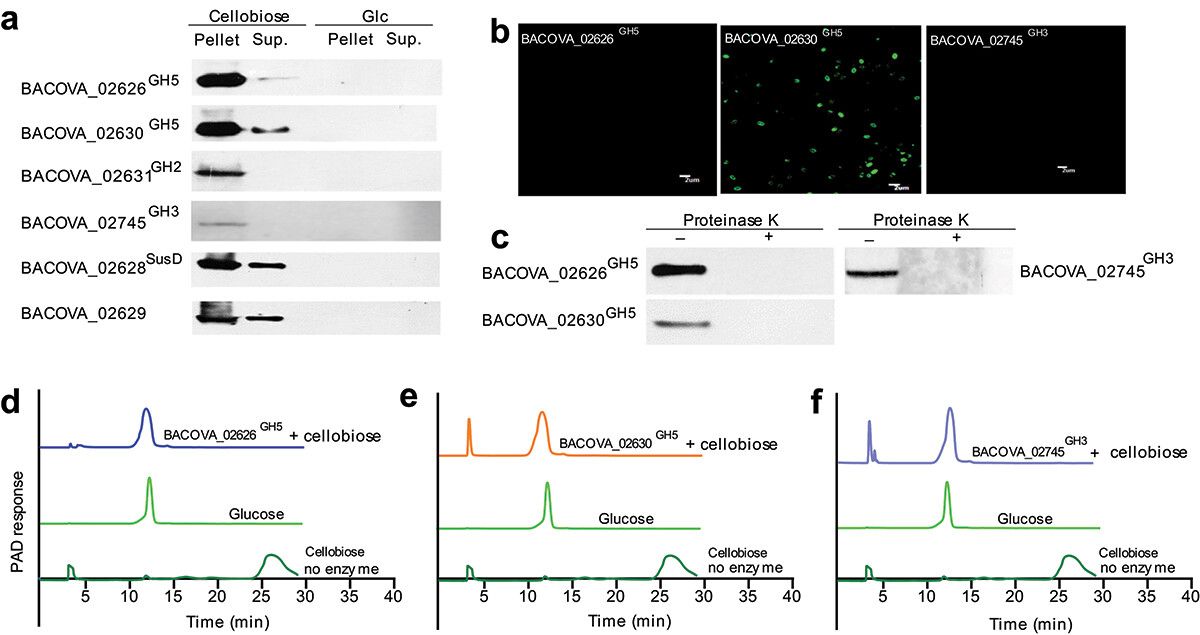 Figure 2. Two new cellulases were determined.
Figure 2. Two new cellulases were determined.
To investigate bacterial functional activities, we used PICRUSt1/2 and Tax4Fun to map 16S rRNA sequences to genes and pathways. Unsupervised clustering of 49 predicted metabolic MetaCyc pathways showed significant differences between the vehicle and cellobiose groups, indicating cellobiose-specific microbiome metabolic pathways. Tax4Fun predicted that only β-N-acetylhexosaminidase, alpha-glucosidase, and β-galactosidase differed from the vehicle group. PICRUSt1 analysis revealed enriched KEGG pathways related to histidine, methane, Vitamin B6, and folate biosynthesis after cellobiose treatment. Histidine metabolism and β-N-acetylhexosaminidase have been shown to inhibit colon inflammation. Cellobiose treatment decreased TNF-α and IL-6 mRNA and protein levels and increased H2R mRNA expression. These findings suggest cellobiose may alter bacterial metabolic functions, though its impact on inflammation remains unclear and requires further investigation.
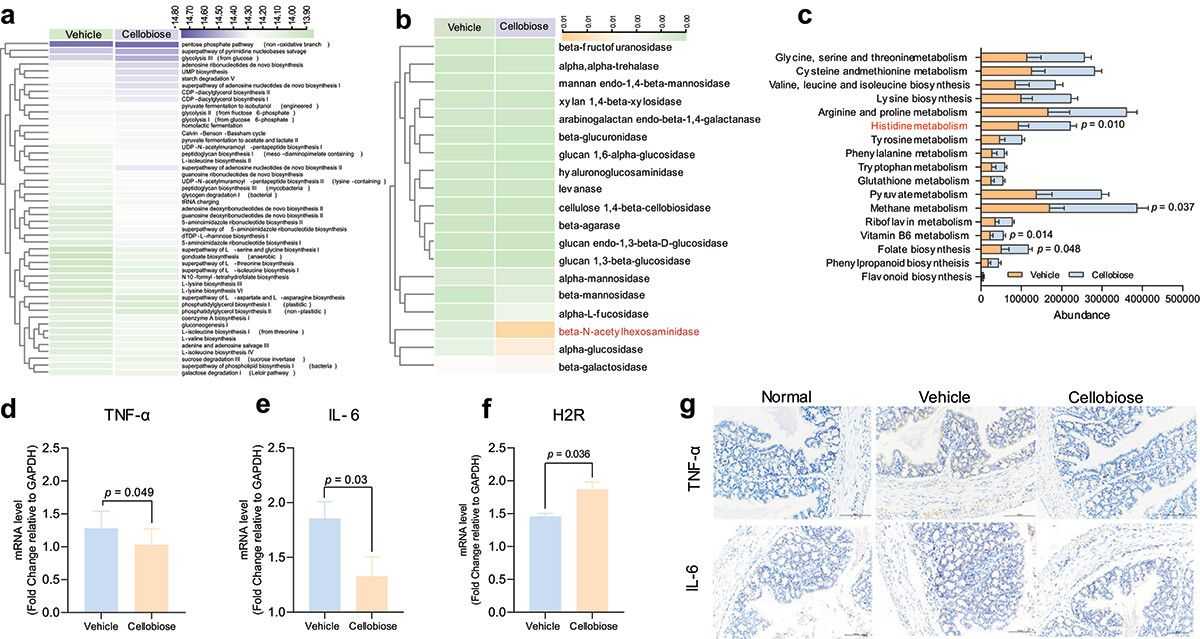 Figure 3. Predication of metabolic function of bacteria and inflammatory factors detection.
Figure 3. Predication of metabolic function of bacteria and inflammatory factors detection.
Conclusion
Cellulolytic bacteria are crucial for cellulose breakdown, yet their molecular mechanisms remain underexplored. Cellobiose, a cellulose unit, promotes the growth of key gut bacterium BO, but its molecular mechanism is unclear. This study identified two new cell surface cellulases in BO that degrade cellobiose into glucose. These cellulases, with structures similar to soil bacteria enzymes, indicate an evolutionary adaptation. In vivo tests showed cellobiose reshaped gut microbiota and reduced inflammation, suggesting potential anti-inflammatory properties. This research enhances our understanding of cellulose degradation in the human gut and highlights the need for further exploration of gut microbiota's cellulolytic capabilities.
Reference
- Li M, Wang Y, Guo C, et al. The claim of primacy of human gut Bacteroides ovatus in dietary cellobiose degradation. Gut Microbes, 2023, 15(1): 2227434.
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


