
 Sample Submission Guidelines
Sample Submission Guidelines
Long-Read Sequencing
CD Genomics offers cutting-edge technologies and bioinformatics analysis services of long-read sequencing, which include PacBio SMRT sequencing and Nanopore sequencing. We offer accurate and cost-effective sequencing solutions for humans, animals, plants, and microbial research.
What is Long-Read Sequencing
Long-read sequencing, also known as third-generation sequencing, is a group of booming DNA sequencing technologies that eliminate the need for traditional sequencing techniques DNA cleavage and amplification and allows for the simultaneous detection of long DNA sequences up to 100,000 base pairs. Scientists have come to realize the significance of structural variants in the human genome. Some of them are insertional variants that exceed the read length of many sequencing technologies; some are repetitive regions that make sequence alignment difficult; and some are GC-rich regions, which often leave many inconveniences in studying structural variants by short-read sequencing methods.
In recent years, novel technologies that can detect long-read sequences have emerged, providing researchers with new strategies and tools for genome analysis. Among them, the most representative and widely used are PacBio SMRT sequencing and Nanopore sequencing. Long-read sequencing technologies can produce very long reads, which is not possible in next-generation sequencing.
PacBio launched the sequencing platform of single molecule real time sequencing (SMRT), which uses a customized flow cell with lots of zero-mode waveguides to sequence single molecules in real-time (ZMW). The polymerase is attached to the well's bottom and enables the DNA strand to pass through the ZMW. Real-time imaging of fluorescently tagged nucleotides synthesized alongside specific DNA template molecules is possible with SMRT sequencing. When the framework and polymerase separate, the sequencing reaction is complete.
The core elements of Nanopore's technology divide into the formation of transmembrane channels from nanopores that allow ionic currents to pass through and the measurement of the changes in current. When molecules such as DNA or RNA pass through nanopores, they cause disruption in the current. The information about the changes in current can be used to identify the molecule. It sequences the whole DNA/RNA molecules directly.
Advantages of Long-Read Sequencing
- Long average read lengths and high consensus accuracy
- Improved accuracy for repeated sequences and copy number variations
- More accurate detection of a large number of mutations
- Optimization of DNA extraction protocol in long-read sequencing
- Compatible with both genome and transcriptome analysis
- Rapid and affordable
Application of Long-Read Sequencing
- Gene function: Focus on samples carrying specific functions to reveal the main causes of different functions.
- Gene structure: Alternative splicing, APA, fusion gene, SSR, CDS prediction, TSS/TES identification, etc.
- Full-length quantification: Find comprehensive and effective differential genes and identify functional genes.
- Epigenomics: Direct full-length genome and transcriptome sequencing can detect base modifications.
Long-Read Sequencing Workflow
CD Genomics employs multiple platforms to provide the fast and accurate Long-Read Sequencing services and bioinformatics analysis. Our highly experienced experts execute quality management, following every procedure to ensure high quality results. The general workflow for Long-Read Sequencing is outlined below.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
|
Analysis Pipeline
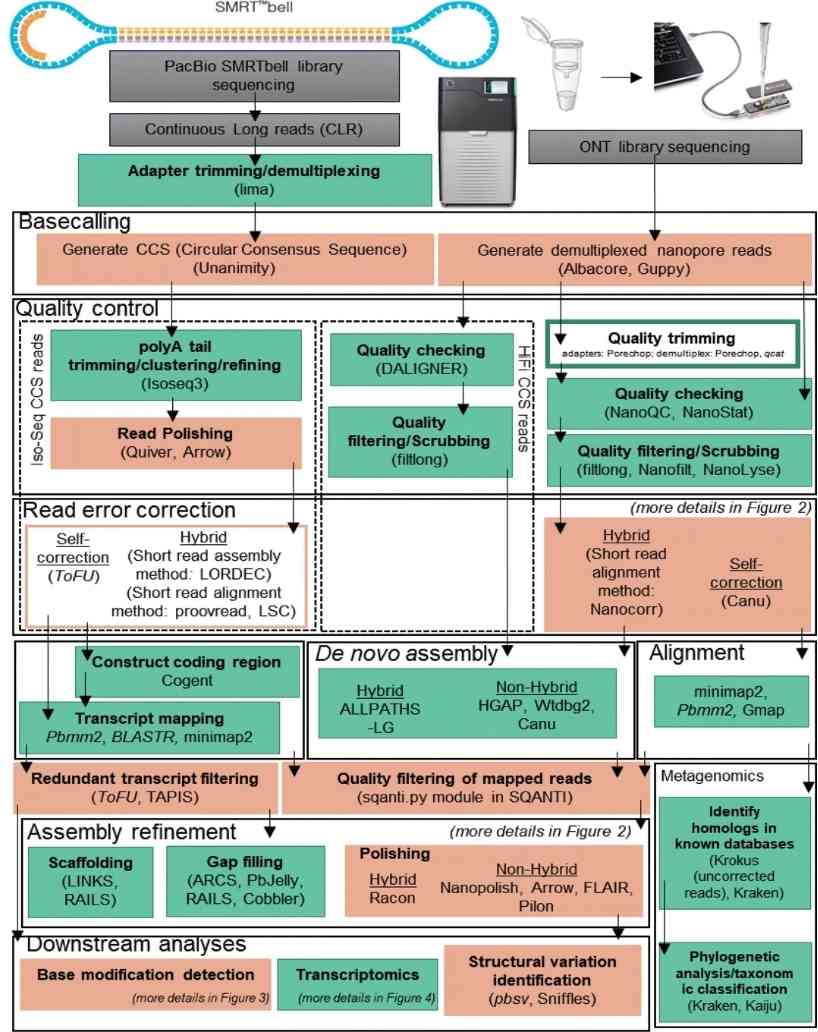 Overview of long-read analysis tools and pipelines. (Amarasinghe et al., 2020)
Overview of long-read analysis tools and pipelines. (Amarasinghe et al., 2020)
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in long-read sequencing for your writing (customization)
CD Genomics provides a comprehensive long-read sequencing service package including sample preparation, library construction, SMRT sequencing and/or Nanopore sequencing, and bioinformatics analysis. We can tailor this pipeline to your research interest. If you have additional requirements or questions, please feel free to contact us.
Reference
- Amarasinghe S L, Su S, Dong X, et al. Opportunities and challenges in long-read sequencing data analysis. Genome biology, 2020, 21(1): 30.
Demo Results
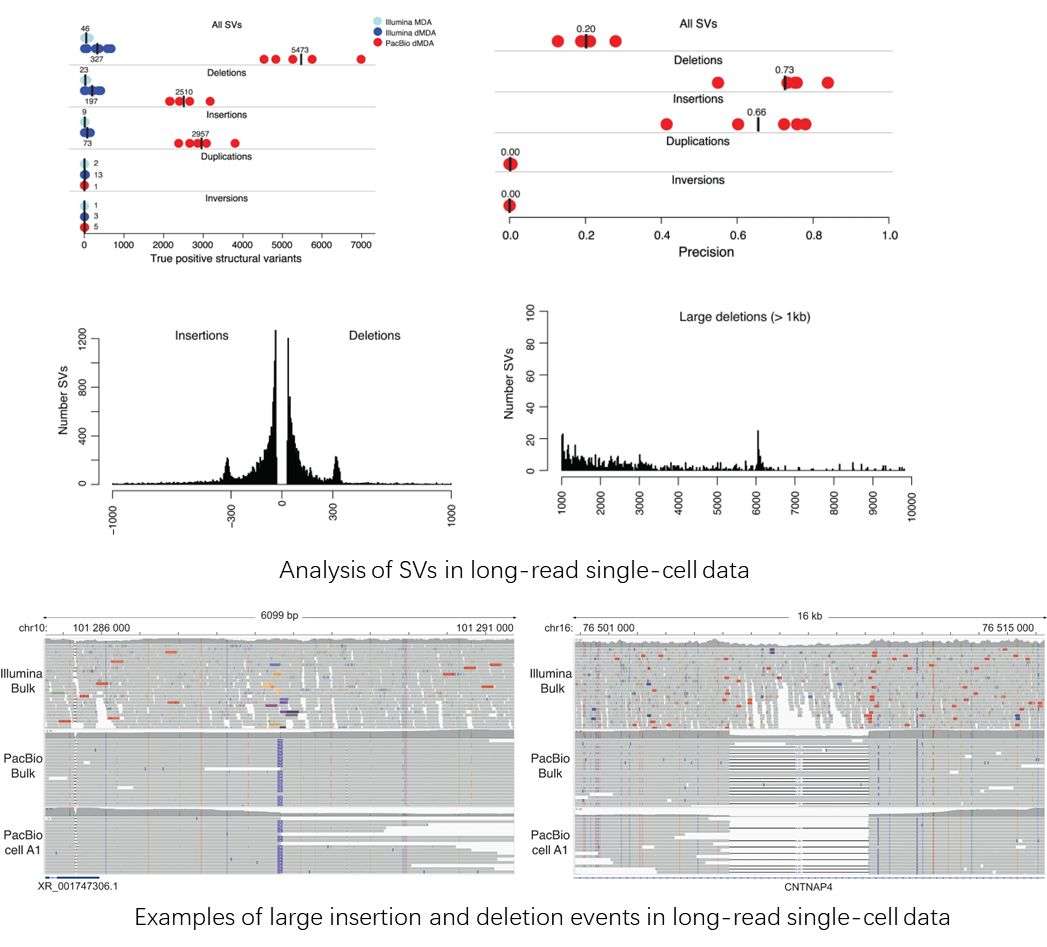 Using single-cell whole-genome long read sequencing results as an example. (Hård et al., 2023)
Using single-cell whole-genome long read sequencing results as an example. (Hård et al., 2023)
Reference
- Hård J, Mold J E, Eisfeldt J, et al. Long-read whole-genome analysis of human single cells[J]. Nature Communications, 2023, 14(1): 5164.
Long-Read Seq FAQs
1. What is the difference between Illumina sequencing and long-read sequencing?
Illumina sequencing, referred to as short-read sequencing, yields shorter sequence reads, typically between 100 and 300 base pairs long. Conversely, long-read sequencing technologies, like those offered by Oxford Nanopore Technologies and Pacific Biosciences, generate significantly longer reads, extending from thousands to tens of thousands of base pairs. This variance in read length enables long-read sequencing to enhance the resolution of intricate genomic regions, including repetitive sequences and structural variants, leading to a more thorough genomic understanding compared to Illumina sequencing. The application of long-read sequencing greatly assists in the deciphering of complex bacterial genomes, particularly when combined with short-read Illumina data through hybrid assembly methods.
2. What research fields is long-read sequencing suitable for?
Long-read sequencing is applicable to various research domains within genomics, including human genetics, evolutionary biology, microbiology, and botany. It enables researchers to gain a more comprehensive understanding of genome structure and function, thereby driving advancements in these fields.
3. What are the advantages and disadvantages of the PacBio platform?
The PacBio platform boasts high single-molecule accuracy, with over 90% of sequences achieving a quality score (Q) of above 30, and offers sequencing data with alignment accuracy surpassing that of NGS platforms. Moreover, it provides an average read length that surpasses NGS platforms by more than two orders of magnitude. However, its drawbacks include significantly higher sequencing costs compared to platforms like Novaseq, high equipment expenses, and the inability to interpret sequencing data in real-time, thus failing to meet the demands of scenarios requiring high timeliness.
4. What are the advantages and disadvantages of ONT technology?
The advantages of ONT technology lie in its high sequencing timeliness and relatively lower sequencing costs compared to the PacBio platform. Moreover, its average read length surpasses that of NGS platforms by more than two orders of magnitude. However, its disadvantages stem from systematic errors in signal detection, such as difficulties in identifying base homopolymers, resulting in significant differences in both single-molecule accuracy and alignment accuracy compared to PacBio and NGS platforms. These limitations restrict its applications.
Long-Read Seq Case Studies
The landscape of genomic structural variation in Indigenous Australians
Journal: Nature
Impact factor: 45.16
Published: 13 December 2023
Background
Australia hosts hundreds of Aboriginal nations, each with rich cultural and linguistic diversity. Despite extensive documentation of their cultural heritage, the genomic diversity of Indigenous Australians is underexplored, with significant under-representation in global genomic databases. The National Centre for Indigenous Genomics (NCIG) addresses this gap by engaging Aboriginal communities in ethical and community-led genomics research. In this study, population-scale long-read sequencing was conducted on Aboriginal communities using Oxford Nanopore Technologies, revealing previously uncharted genomic variations and structural variants, which are crucial for advancing genomic medicine.
Methods
Sample Preparation:
Saliva sample collection
Fresh blood collection
DNA extraction
Sequencing:
Library construction
Whole-genome ONT sequencing
ONT data processing
Alignment to reference genome
Detection of structural variation
Structural variation repeat classification
Comparison to annotations
Analysis of large CNVs
Results
The complete T2T-chm13 genome reference was used to evaluate mappability and structural variant (SV) detection, showing ONT's superior alignment and SV detection. This revealed 159,912 unique SVs and 136,797 large indels, and identified 11 high-confidence copy number variants (CNVs) per individual, highlighting the benefits of long-read sequencing for detailed genomic profiling.
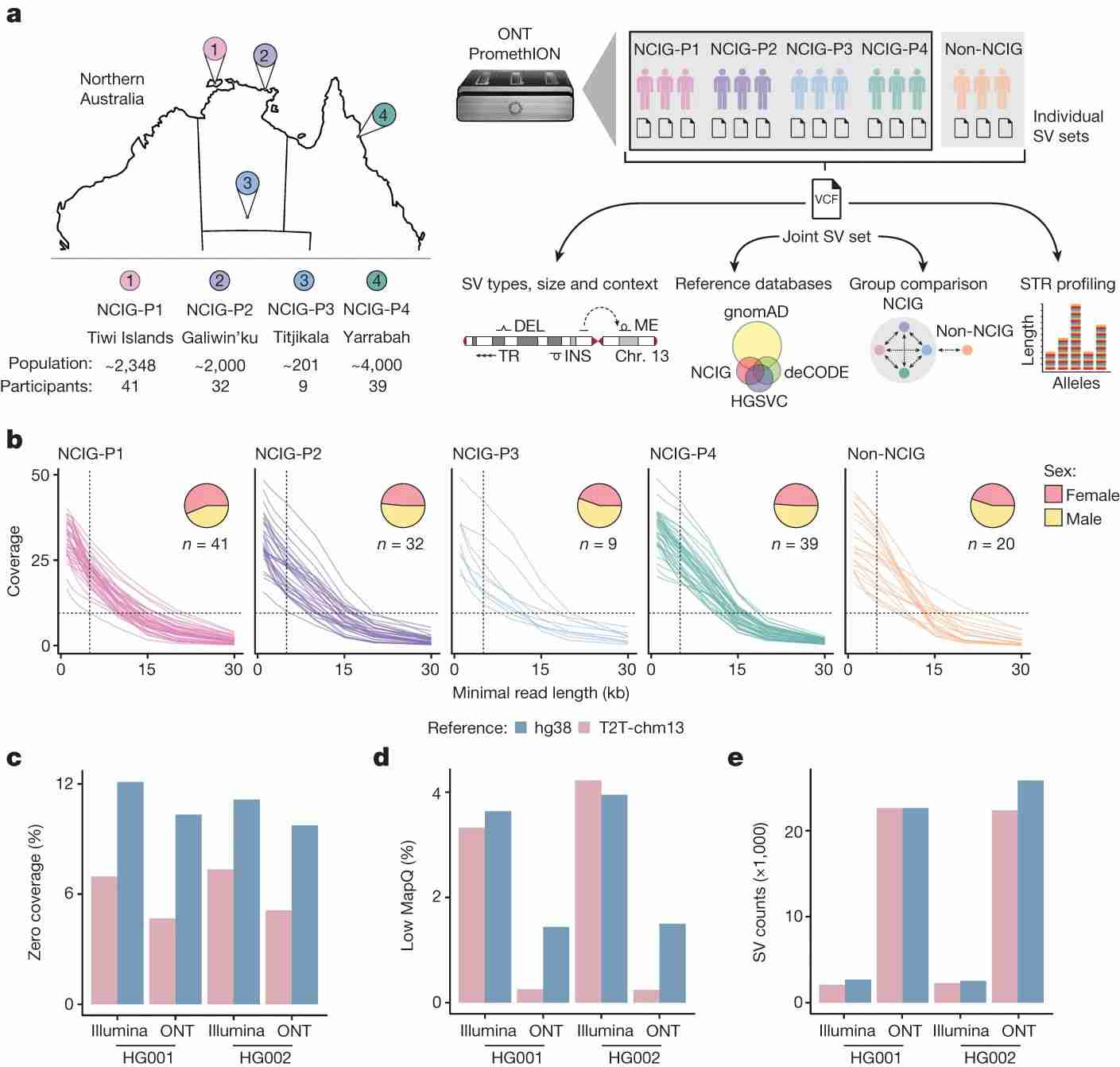 Fig. 1 Long-read sequencing in Indigenous Australian communities.
Fig. 1 Long-read sequencing in Indigenous Australian communities.
To characterize genomic structural variation, the authors categorized variants by type, size, and context. Most variants (84.9%) were repetitive sequences, including STRs, TRs, and mobile element insertions/deletions. CNVs were fewer but covered significant genome regions. Variants were unevenly distributed, with higher density near telomeres, particularly TR-associated SVs. Size varied by type, with TR-SVs larger than STRs and non-repetitive SVs. Mobile element SVs showed size peaks corresponding to major repeat families like Alu, L1, and SVA. Many SVs were unique to this study, especially given the inclusion of under-represented Australian communities and the use of long-read sequencing. Comparing this study's SVs to global databases, the authors found around 38% similarity with annotated SVs, indicating a high proportion of novel SVs.
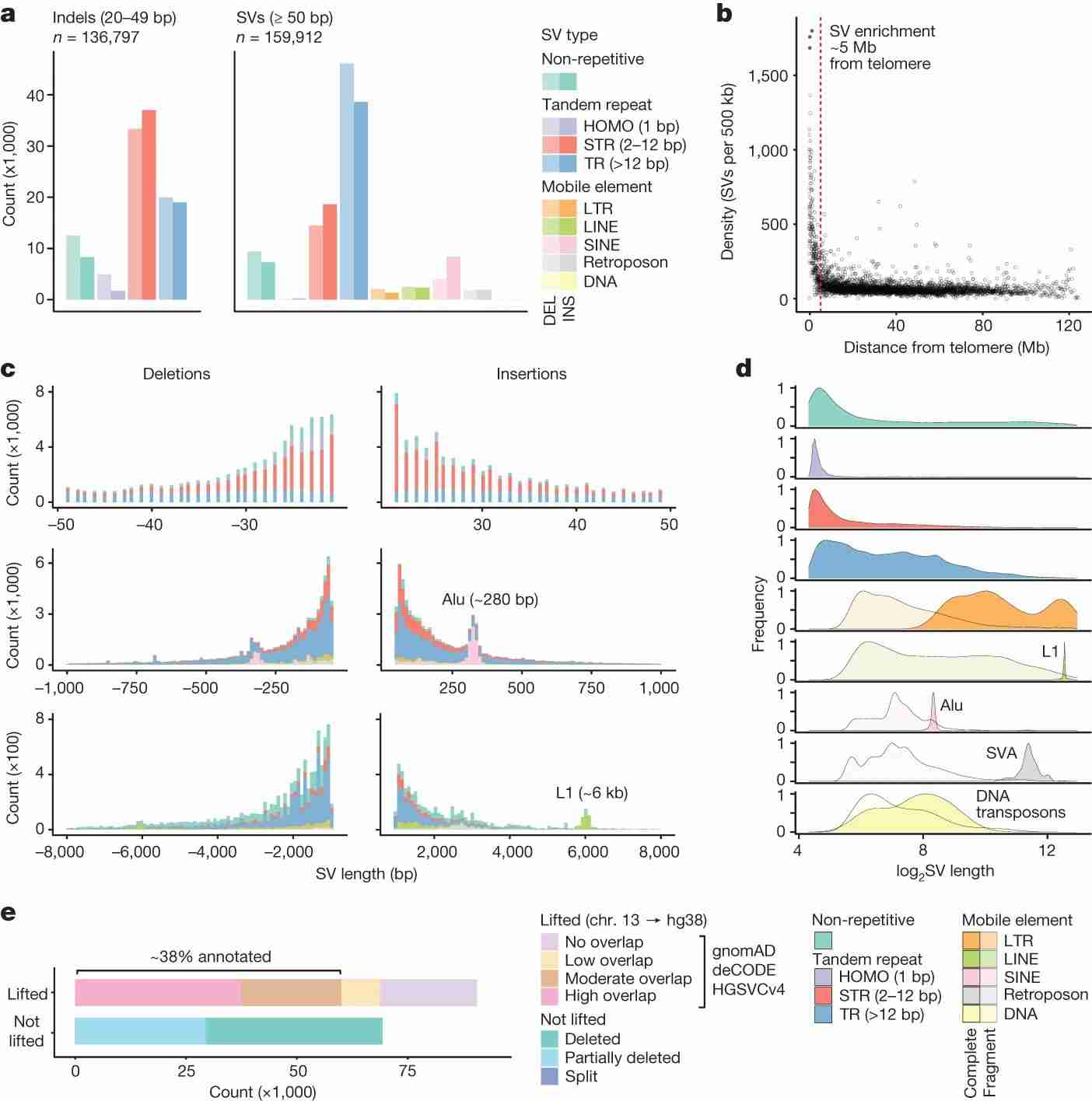 Fig. 2 Landscape of genomic structural variation.
Fig. 2 Landscape of genomic structural variation.
The authors analyzed genomic structural variations (SVs) among Indigenous and non-Indigenous Australians, finding most SVs to be private or polymorphic. Notably, 48.5% of SVs were unique to Indigenous individuals, while 9.2% were unique to non-Indigenous individuals. Unique SVs in Indigenous individuals were often unannotated and specific to particular communities. Genetic distinctions were evident, with distinct clusters for different communities in principal coordinate analysis, and shared SVs among Indigenous individuals were rare, underscoring their unique genomic diversity.
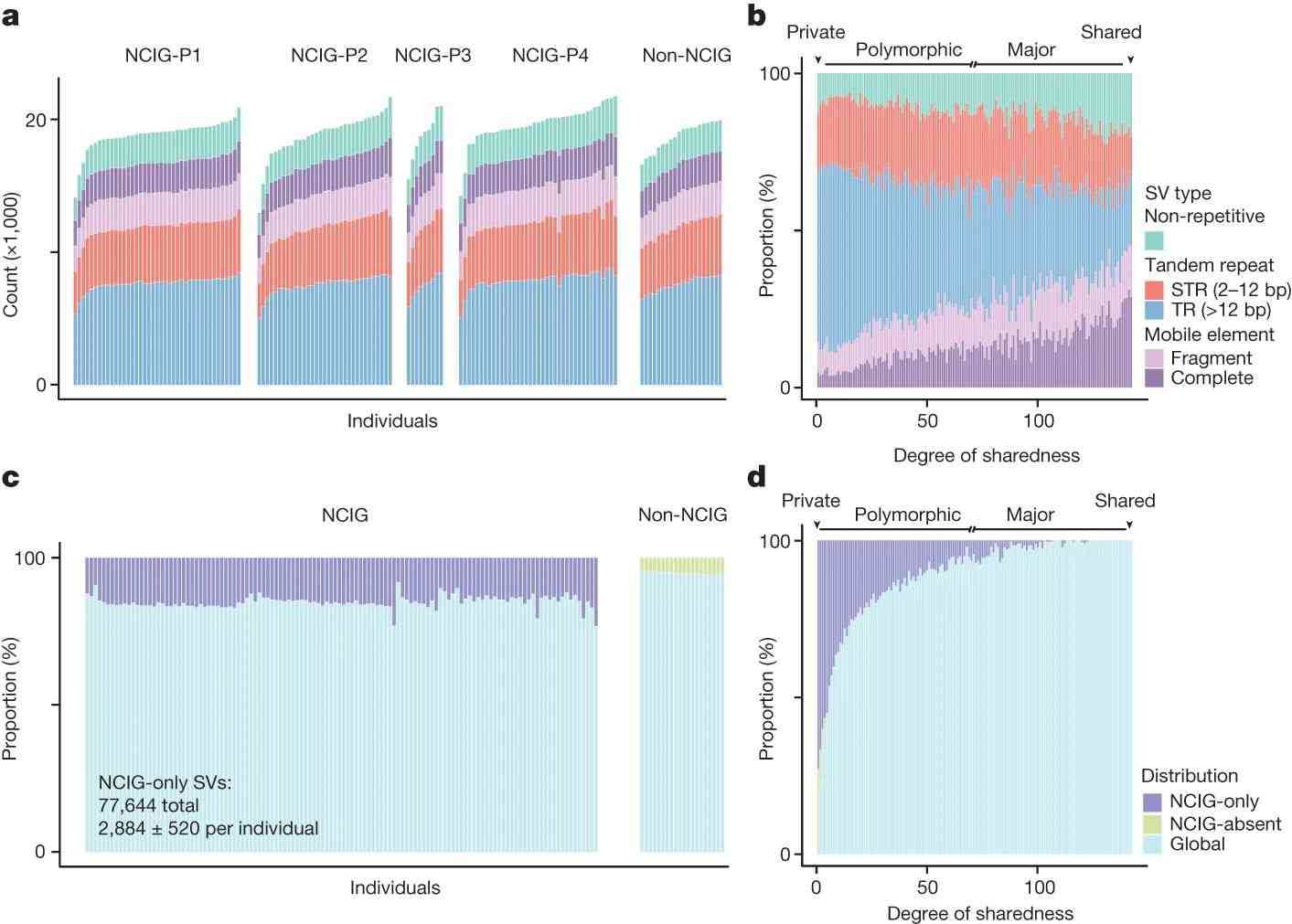 Fig. 3 Distribution of SVs in Indigenous and non-Indigenous individuals.
Fig. 3 Distribution of SVs in Indigenous and non-Indigenous individuals.
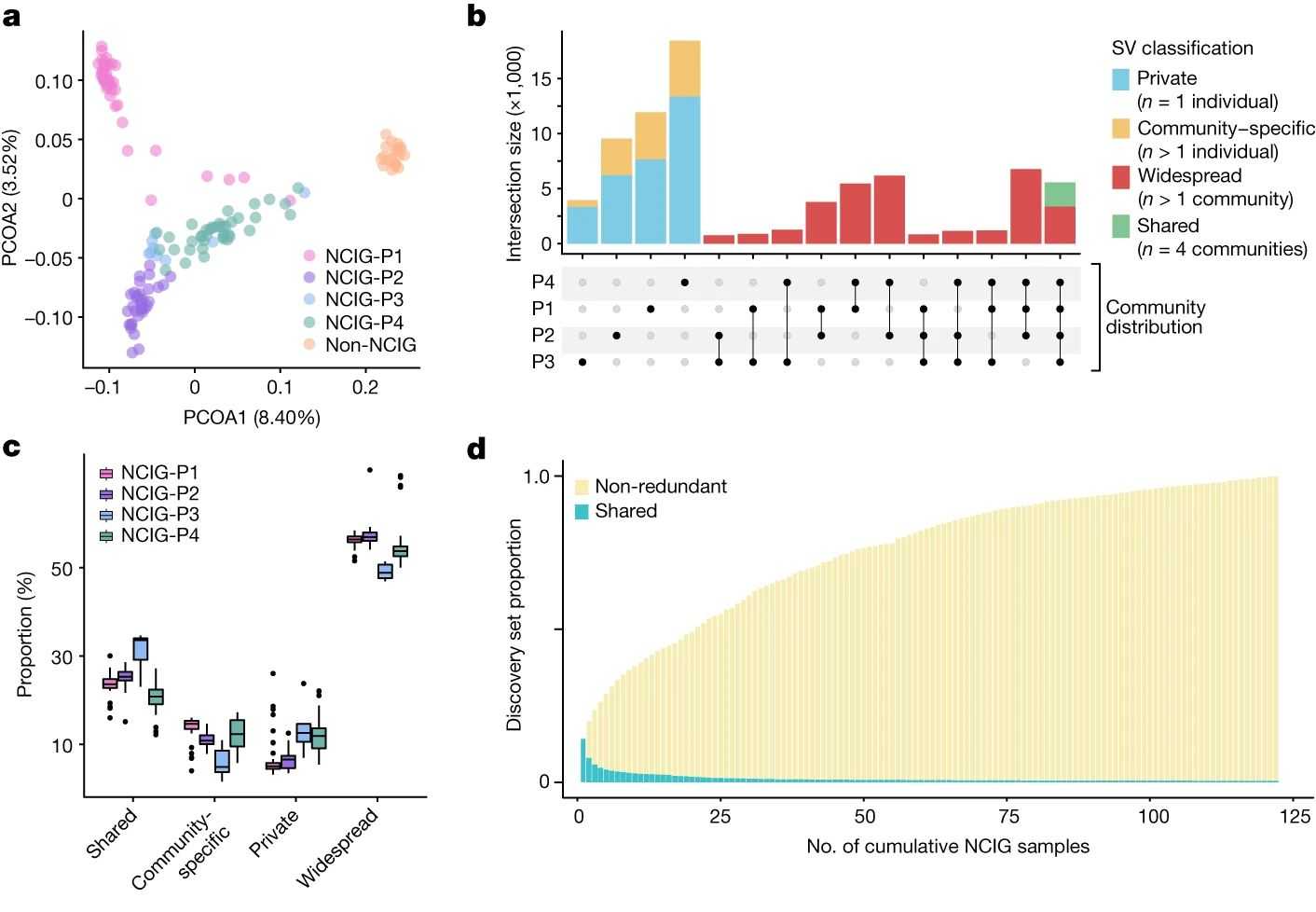 Fig. 4 Distribution of SVs between Indigenous communities.
Fig. 4 Distribution of SVs between Indigenous communities.
Conclusion
This study unveils the structural variation landscape of Indigenous Australians using long-read sequencing, revealing diverse variations unique to this population. This underscores the need for ancestry-specific reference data in genomic medicine. We highlight the genetic heterogeneity among Indigenous communities and provide insights into broader patterns of genomic structural variation in human populations, particularly in short tandem repeats (STRs), which have implications for disease diagnosis and treatment.
Reference
- Reis, A.L.M., Rapadas, M., Hammond, J.M. et al. The landscape of genomic structural variation in Indigenous Australians. Nature 624, 602–610 (2023).
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Journal: Cell Death & Disease
Year: 2024
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
See more articles published by our clients.


