
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics has extensive experience in offering Iso-Seq service by producing full-length transcripts without assembly. Strict quality control following every procedure is executed to ensure comprehensive and accurate results.
What is Iso-Seq
Isoform Sequencing (Iso-Seq) is a high-throughput sequencing technology developed by PacBio, leveraging Single Molecule, Real-Time (SMRT) technology. Unlike fragmented RNA sequencing methods that require reassembly, Iso-Seq directly sequences full-length cDNA transcripts, encompassing the complete 5' untranslated region (5' UTR) through to the 3' polyadenylation (polyA) tail. This approach allows for a more accurate analysis of the target gene set or the entire transcriptome, providing insights that are otherwise challenging to obtain through traditional Next-Generation Sequencing (NGS) techniques.
The Introduction of Iso-Seq
In eukaryotic organisms, a single gene may encode a surprising number of proteins after alternatively spliced, each with a distinct biological function. There are known human genes that have very different functions depending on which splice variant is expressed. With alternative splicing so critical to genome function.
Short-read RNA-Seq works by physically shearing transcript isoforms into smaller pieces and reassembling, leaving possibilities for mis-assembly or incomplete capture of the full diversity of isoforms from genes of interest. It's important to capture full-length transcripts. By taking advantage of PacBio SMRT long reads sequencing technology, the isoform sequencing (Iso-Seq) can easily cover complete transcript from the 5' end to the 3'-poly A tail without the need of fragmentation to obtain full-length cDNA sequences, which is useful to identify new transcripts and new introns, thus accurately identifying isoforms, alternative splicing sites, fusion gene expression, and allelic expression.
Comparing with Illumina platform, PacBio analysis can easily detect very long polycistronic RNA molecules (called complex transcripts), and reveal a wide variety of novel transcriptional overlaps between adjacent and distant genes situated in parallel. It is helpful to raise the possibility to study a genome-wide network exerting joint control on gene expression and replication.
We also provide Nanopore Full-Length Transcripts Sequencing services. For further details, please visit the relevant page.
Key Features and Advantages of Iso-Seq
- Direct acquisition of full-length transcript sequences, enabling a more accurate reflection of the transcriptome information for the sequenced species.
- Detection of multiple alternative splicing forms, uncovering additional splice sites and alternative splicing events.
- Discovery of novel functional genes, augmenting genome annotation.
- Facilitation of precise analysis of fusion genes, homologous genes, gene superfamilies, or alleles.
- High accuracy, continuity, and completeness of transcript sequences.
- Integrated with second-generation transcriptome sequencing, enabling precise quantification at both gene and transcript levels.
- High species adaptability, allowing analysis with or without a reference genome.
Applications of Iso-Seq
- Genomic Annotation: Conduct precise annotation of new genomes, delivering high-quality transcript information.
- Transcriptome Analysis: Investigate the diversity of transcripts across various tissues or cell types, elucidating the complexity of gene expression.
- Disease Research: Identify specific gene fusions and alternative splicing events in cancer and genetic disease studies.
- Agricultural Biotechnology: Apply in crop genomics research to enhance breeding strategies for improved crop varieties.
Iso-Seq Workflow
CD Genomics utilizes PacBio SMRT technology for Full-Length Transcript Sequencing (Iso-Seq). This advanced method involves extracting high-quality RNA, synthesizing full-length cDNA, constructing cDNA libraries, and performing high-fidelity sequencing to capture complete transcript sequences, ensuring comprehensive and accurate transcriptome analysis.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses: Transcriptome with no Reference Standard data analysis
Standard data analysis
|
Analysis Pipeline
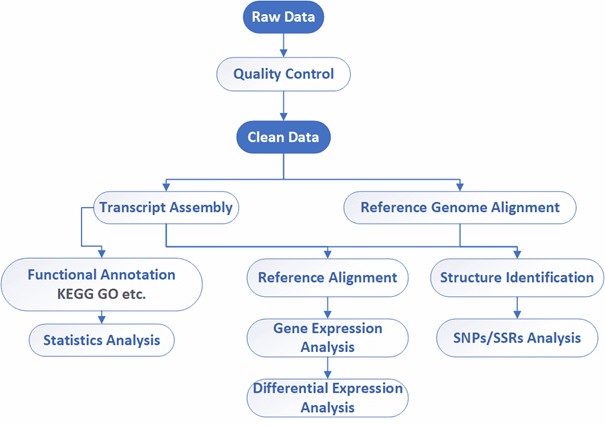
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Iso-Seq for your writing (customization)
Long-read sequencing allows the straightforward identification of alternatively transcribed or processed transcripts, polycistronic transcription units, and other long cDNA sequences. With expertise and dedication, advanced PacBio SMRT platform and services from CD Genomics will be your best companion in Iso-Seq. Please contact us for more information and a detailed quote.
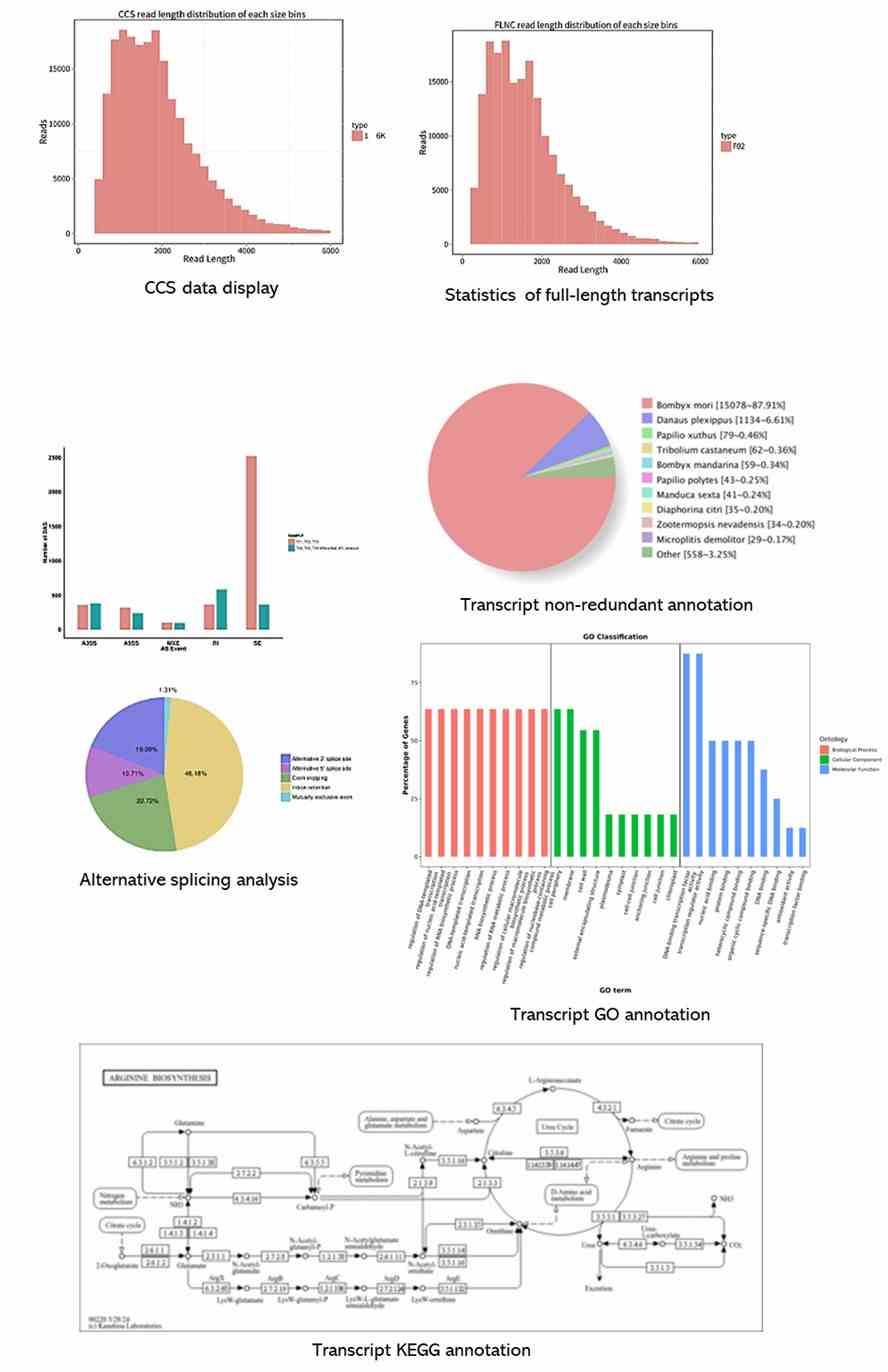
Partial results are shown below:
1. In the field of transcriptomics, what do isoform and transcript refer to, and what are their differences?
An isoform denotes RNA molecules that are transcribed from the same gene locus but exhibit structural variations. In contrast, an RNA transcript is an RNA molecule copy produced during gene transcription. Essentially, both terms describe the same entity-RNA molecules. However, the emphasis differs: isoform highlights structural differences in RNA molecules, such as TSS, CDS, and UTR regions, whereas transcript focuses on the post-transcriptional processing the RNA molecules have undergone, such as which exons have been spliced together.
2. What are the Characteristics of Transcripts Targeted by Iso-Seq Sequencing?
- Typical Structure: Eukaryotic mRNA transcripts usually consist of a 5' cap, 5' UTR, CDS, 3' UTR, and polyA tail. Iso-Seq leverages polyA or specific sequences to enrich mRNA or target specific gene RNAs.
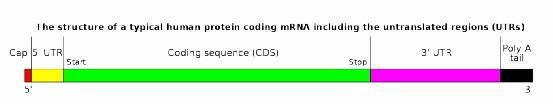 Figure 1 Typical mRNA Structure.
Figure 1 Typical mRNA Structure.
- Length Distribution: Transcript lengths predominantly range from 1 to 3 kilobases. For instance, over 99% of human transcripts are shorter than 10 kilobases, which allows Iso-Seq to easily achieve full-length sequencing.
- Complexity: Following transcription, a gene can undergo alternative splicing to produce multiple transcripts with different biological functions. Accurately distinguishing these fine structural differences between transcripts has posed significant challenges to techniques preceding Iso-Seq.
3. What are the types of SMRTbell designs for PacBio-supported Iso-Seq libraries?
Currently, PacBio-supported Iso-Seq sequencing libraries include several types of SMRTbell designs:
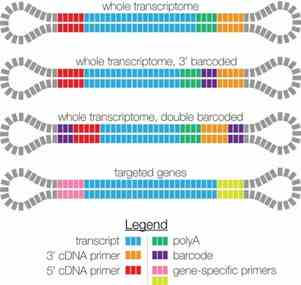 Figure 2 SMRTbell Design.
Figure 2 SMRTbell Design.
These designs allow for flexible applications: by optionally adding barcodes, Iso-Seq supports both single-sample and multiplexed sequencing; by using different primers for reverse transcription, it facilitates both whole-transcriptome sequencing and targeted sequencing of specific gene sets.
4. What basic steps are involved in the data preprocessing of Iso-seq, and what are their objectives?
Iso-Seq data preprocessing is achieved through SMRT tools components ccs, lima, and IsoSeq3. The steps include:
- Correcting random sequencing errors in subreads under the ccs sequencing mode to obtain high-quality HiFi reads.
- (For multiplexed sequencing) Splitting samples using barcodes and removing barcode primer sequences.
- Identifying and removing polyA tails and concatemers.
- Clustering and deduplicating reads to generate consensus sequences.
Utilizing PacBio Iso-Seq for Novel Transcript and Gene Discovery of Abiotic Stress Responses in Oryza sativa L.
Journal: International Journal of Molecular Sciences
Impact factor: 6.208
Published: 31 October 2020
Background
Global climate change exacerbates abiotic stress conditions, negatively impacting crop yields. Rice, a staple for over half the world's population, has extensive genetic diversity. However, current studies rely on the japonica reference genome, potentially missing crucial genetic information from other subspecies. To address this, the authors used PacBio SMRT Iso-Seq to sequence and reconstruct partial transcriptomes from various rice subspecies, enabling the discovery of novel genes for stress tolerance without needing an existing reference genome.
Materials & Methods
Sample Preparation
- Plant material
- Rice
- RNA extraction
Sequencing
- RNA-Seq
- De novo transcriptome reconstruction
- PacBio Iso-Seq
- InDel analysis
- BUSCO analysis
- Phylogenetic analysis
- Functional annotation
- Differential gene expression analysis
Results
The authors selected ten rice cultivars, isolating RNA from plants grown under diverse conditions. Sequencing on the PacBio platform yielded 15.49 to 24.51 gigabases per cultivar. Using IsoSeq3, they obtained 37,535 to 54,594 high-quality full-length transcripts per cultivar after filtering for plant-specific sequences and removing contaminants.
Table 1. Sampling for PacBio isoform sequencing.
![]()
During library preparation, degraded 5′ RNA products can lead to redundant isoforms lacking full 5′ sequence information. Three methods-cogent, cDNA cupcake, and TAMA-were tested to collapse these redundancies. While cDNA cupcake and TAMA use a reference genome, cogent reconstructs a coding genome for collapsing. All methods significantly reduced isoform numbers, with cogent showing more unmatched transcripts. Post-collapsing, transcript lengths increased and isoform uniqueness improved, particularly with TAMA having the highest proportion of unique isoforms per gene locus. Phylogenetic analysis based on SNPs highlighted distinct clusters by subspecies, emphasizing genetic differences among aus, indica, and japonica cultivars.
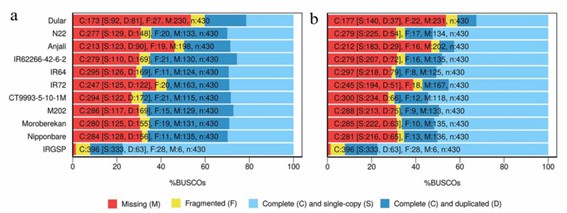 Figure 1. BUSCO assessment analysis of uncollapsed (a) and collapsed (b) transcripts.
Figure 1. BUSCO assessment analysis of uncollapsed (a) and collapsed (b) transcripts.
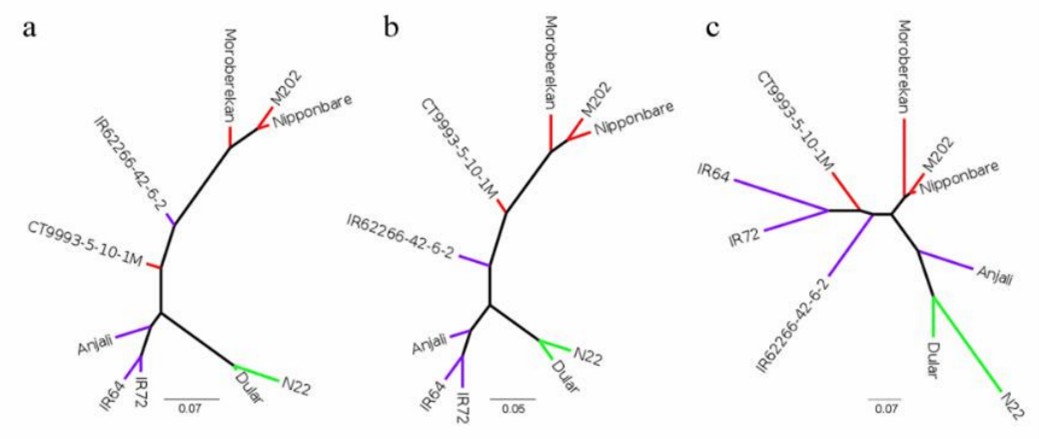 Figure 2. Phylogenetic trees constructed with SNPhylo.
Figure 2. Phylogenetic trees constructed with SNPhylo.
The study used TAMA to collapse HQ transcripts and cogent for unmapped ones, resulting in 10,511 to 15,011 gene loci and 14,255 to 20,803 unique isoform models per cultivar. About one third of gene loci and half of transcript models matched the Nipponbare reference. Functional annotation revealed 60% to 70% complete ORFs and highlighted major biological processes. However, a notable portion of transcripts remained unannotated, potentially originating from wild Oryza species.
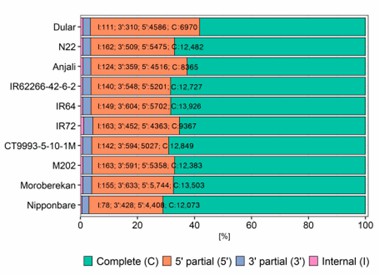 Figure 3. Fraction of predicted open reading frames (ORFs) using TransDecoder.
Figure 3. Fraction of predicted open reading frames (ORFs) using TransDecoder.
To identify common and specific transcripts among cultivars, one cultivar from each subspecies (N22, IR64, Nipponbare) was used as a blast database. Around 9,000 transcripts were common across all cultivars, with 652 unique to N22 and 2426 to IR64. Differential gene expression analysis in N22 revealed 56 aus-specific genes responsive to combined drought and heat stress, including the upregulated B12288 gene, which shares homology with drought-responsive genes in other Oryza species and Arabidopsis thaliana.
 Figure 4. Multiple sequence alignment of five Oryza RAB21 dehydrin proteins.
Figure 4. Multiple sequence alignment of five Oryza RAB21 dehydrin proteins.
Conclusion
The authors sequenced transcriptomes of ten rice cultivars using PacBio Sequel, yielding 37,500 to 54,600 high-quality isoforms per cultivar. Their de novo reconstructions included about 40% novel isoforms compared to Nipponbare. For the drought/heat tolerant aus cultivar N22, they identified 56 differentially expressed genes in developing seeds under field conditions, offering a cost-effective way to identify stress tolerance genes absent in standard genome assemblies.
Reference
- Schaarschmidt S, Fischer A, Lawas LM, et al. Utilizing PacBio iso-seq for novel transcript and gene discovery of abiotic stress responses in Oryza sativa L. International Journal of Molecular Sciences. 2020, 21(21):8148.
Here are some publications that have been successfully published using our services or other related services:
Bacterial communities of Cassiopea in the Florida Keys share major bacterial taxa with coral microbiomes
Journal: bioRxiv
Year: 2024
Production of a Bacteriocin Like Protein PEG 446 from Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotics and Antimicrobial Proteins
Year: 2024
Untangling the Role of Pathobionts from Bacteroides Species in Inflammatory Bowel Diseases
Journal: bioRxiv
Year: 2023
A chromosome-level genome resource for studying virulence mechanisms and evolution of the coffee rust pathogen Hemileia vastatrix
Journal: bioRxiv
Year: 2022
Streptomyces buecherae sp. nov., an actinomycete isolated from multiple bat species
Journal: Antonie Van Leeuwenhoek
Year: 2020
See more articles published by our clients.


