
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics offers advanced RIP-seq services utilizing next-generation sequencing (NGS) technology. Our RIP-seq service provides precise and comprehensive analysis of RNA-protein interactions, delivering detailed insights into RNA-binding protein interactions across various biological samples.
The Introduction to RIP-Seq
Every activity of transcript lifecycle, from birth (polymerases) to degradation (nucleases), involves protein binding. In addition to protein production, a subset of these transcripts plays crucial roles in other essential processes such as epigenetic regulation and genome protection by transposon silencing. Most studies have focused on transcriptomics profiling. It is believed, however, that the levels of mRNAs do not always directly correlate with the steady-state protein levels. Interest in identifying the RNAs associated with RNA binding protein (RBP) in a cellular context is growing as the role of RNA processing and translational events that occur post-transcriptionally is begin to be appreciated.
RIP-Seq maps the sites at which proteins are bound to the RNA within RNA-protein complexes. RNA immunoprecipitation (RIP) implies the purification of RNA–protein interactions in native conditions by using a protein-specific antibody to map the RBP of interest. The advent of sequencing technologies, coupled to various RIP chemistries, has enabled the simultaneous detection of thousands of bound transcripts (mRNAs, non-coding RNAs or viral RNAs) in a single experiment. CD genomics provides RIPed RNA sequencing to obtain insights into not just the well-established processes such as transcription, splicing and translation, but also in newer fields such as RNA interference and gene regulation by non-coding RNAs.
Advantages of Our RIP-Seq Service
- In current biology research, RIP-seq stands out as the most effective method for determining protein-RNA interactions within the natural state of a cell. This technique efficiently identifies whether a protein is an RNA-binding protein, elucidates which RNAs directly interact with the protein, and pinpoints their binding sites.
- RIP-seq enables the comprehensive investigation of protein-RNA interactions at the whole transcriptome level, shedding light on the types of RNAs involved in these interactions.
- With its high resolution, RIP-seq offers the ability to discern RNA sequences that interact with proteins, ultimately providing valuable insights into the intricate landscape of protein-RNA interactions.
- Cost-Effectiveness: The experimental cycle is short, the analysis is comprehensive, and the price is low.
- High Coverage: The entire transcriptome is covered, allowing for screening and identification of protein-binding sites across the whole transcriptome.
- High Sensitivity: Millions of sequence tags can be obtained from each sample, enabling the discovery of rare protein-binding sites on the transcriptome.
- High Precision: High signal-to-noise ratio data can be obtained, accurately distinguishing true events from noise and precisely locating protein-binding sites.
- Customized RNA Immunoprecipitation Sequencing Plan: Tailored RNA immunoprecipitation sequencing plans can be designed based on specific needs.
Applications of RIP-Seq
- Investigating the Interactions between RNA and Proteins within Cells
- Identifying the Interactions between RBPs and Non-coding RNAs (such as LncRNAs, miRNAs, etc.)
- Mapping the Genome-wide Interactions between RNA and RBPs
RIP-Seq workflow
CD Genomics applies the Illumina NGS equipment to sequence the bound transcripts, unveiling genome-wide interactions of RBPs. Researchers will submit the RNA pulled by the appropriate RIP methods based on specific protein immunoprecipitation from cell lines or tissues. Our RIP-Seq service, offering the workflow from sample QC through data analysis, enables rapid profiling and deep insight into the RNA.
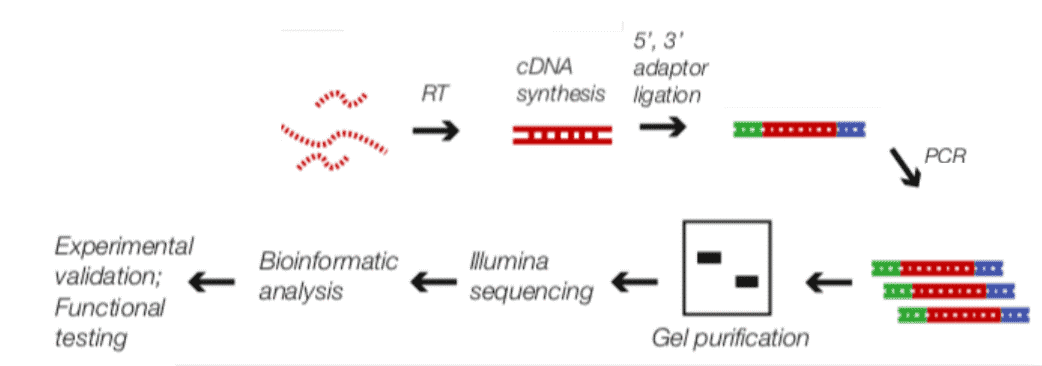 Fig2. RIP-Seq Workflow
Fig2. RIP-Seq Workflow
Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in RIP-Seq for your writing (customization)
If you have additional requirements or questions, please feel free to contact us.
Reference
- Zhao J et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Molecular Cell, 2010 Dec 22;40(6):939-53
Partial results are shown below:
1. What are some recent advances in RIP-seq?
The recent advancements in RIP-seq have evolved significantly. These developments encompass enhancements in antibody engineering focusing on specificity, refined crosslinking techniques aiding in superior RNA-protein complex isolation, and the introduction of sophisticated bioinformatics resources for data interpretation. Moreover, the integration of RIP-seq with methodologies such as CLIP-seq (crosslinking and immunoprecipitation sequencing) offers supplementary perspectives on RNA-protein interactions.
2. How can the success of RIP-seq experiments be assured?
Antibody Validation: Utilize well-characterized, high-affinity antibodies.
Optimized Methods: Adhere to optimized procedures for cell lysis, immunoprecipitation, and RNA extraction.
Proper Controls: Integrate suitable controls (such as non-specific IgG controls, input RNA) to discern specific interactions from background signals.
Replication: Conduct biological replicates to confirm result reproducibility.
Data Integrity: Employ high-quality sequencing platforms and thorough bioinformatics analyses to uphold data accuracy and reliability.
3. What is Input and its role in experiments?
Following RNA fragmentation, prior to immunoprecipitation, a portion of the sample needs to be set aside as Input control (not subjected to immunoprecipitation). The Input consists of the fragmented RNA, which, along with the immunoprecipitated sample RNA, undergoes reverse cross-linking, RNA purification, and subsequent PCR or other detection methods. Through subsequent data analysis, the Input control allows the exclusion of background noise (false-positive peaks caused by nonspecific binding), validates the RNA fragmentation efficacy, and confirms the IP efficiency throughout the experiment. Therefore, the Input control is an indispensable step in IP-seq experiments.
RIP-Seq of EZH2 Identifies TCONS-00036665 as a Regulator of Myogenesis in Pigs
Journal: Frontiers in Cell and Developmental Biology
Impact factor: 6.081
Published: 12 January 2021
Background
Vertebrate skeletal muscle myogenesis involves progenitor cells regulated by Pax3 and Pax7, which induce Myf5 and MyoD for muscle differentiation. Satellite cells aid in regeneration. EZH2, a Polycomb protein, is crucial for muscle growth and gene regulation via histone methylation. lncRNAs interact with EZH2 to regulate myogenesis. This study employed RIP-Seq combined with lincRNA sequencing (lincRNAseq) to identify 356 novel lincRNAs in pig skeletal muscle, revealing insights into their roles, such as TCONS-00036665, which promotes satellite cell proliferation but inhibits differentiation, thus influencing muscle development.
Materials & Methods
Sample Preparation
- Pure Large White female pigs
- Longissimus dorsi muscle tissues
- C57 mice
- Total RNA extraction
Sequencing
- RIP-Seq
- LincRNAseq
- ChIP-Seq
- Illumina HiSeq 2000
- Quality control
- Alignment
- Transcript assembly
- Expression analysis
- Statistical analysis
Results
In this study, RIP-Seq identified EZH2-binding lincRNAs from skeletal muscle of 1-month-old pigs. Using EZH2 antibody enrichment validated by Western blotting, the authors conducted RIP-Seq and lincRNAseq. Filtering steps based on expression levels and CPC values identified 356 EZH2-binding lincRNAs, predominantly located in intergenic regions, suggesting their regulatory roles in muscle biology.
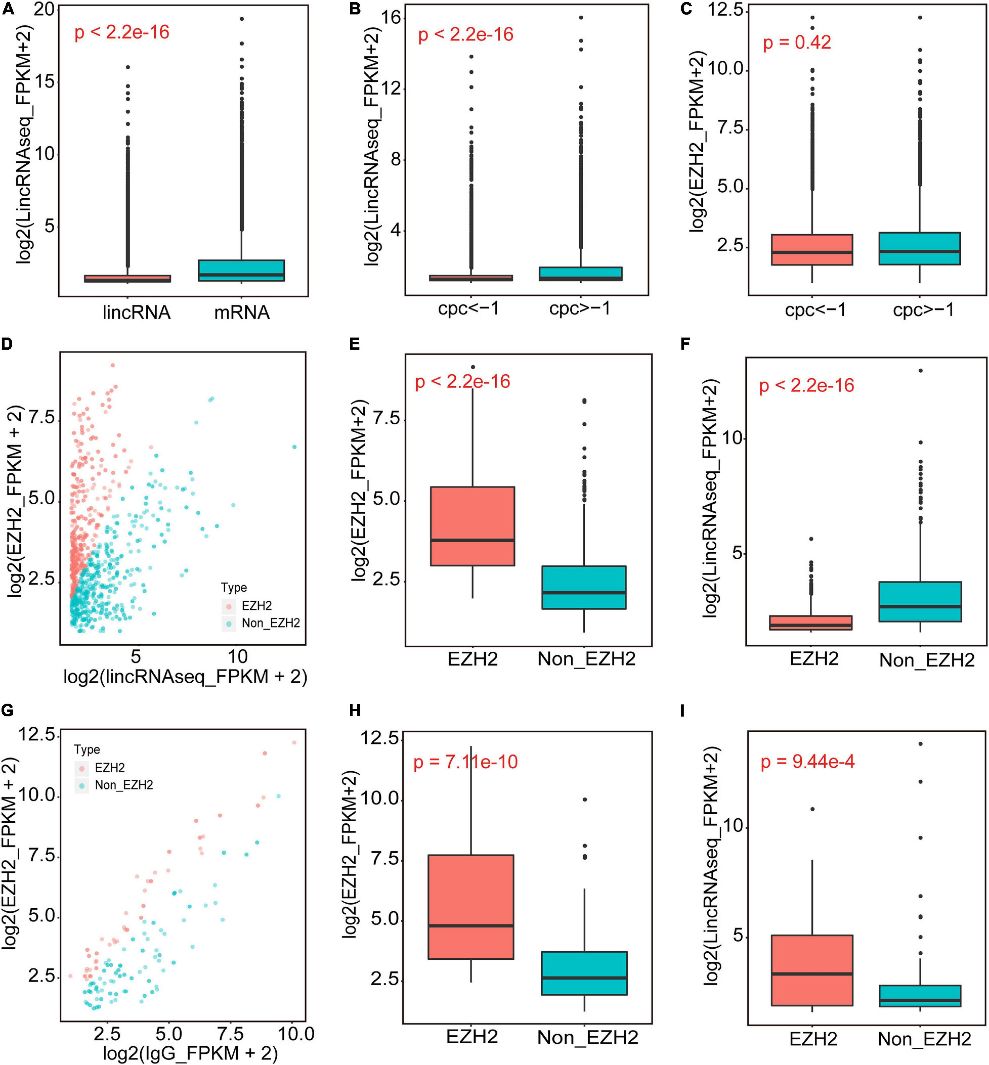 Figure 1. The identification of EZH2-binding novel lincRNAs.
Figure 1. The identification of EZH2-binding novel lincRNAs.
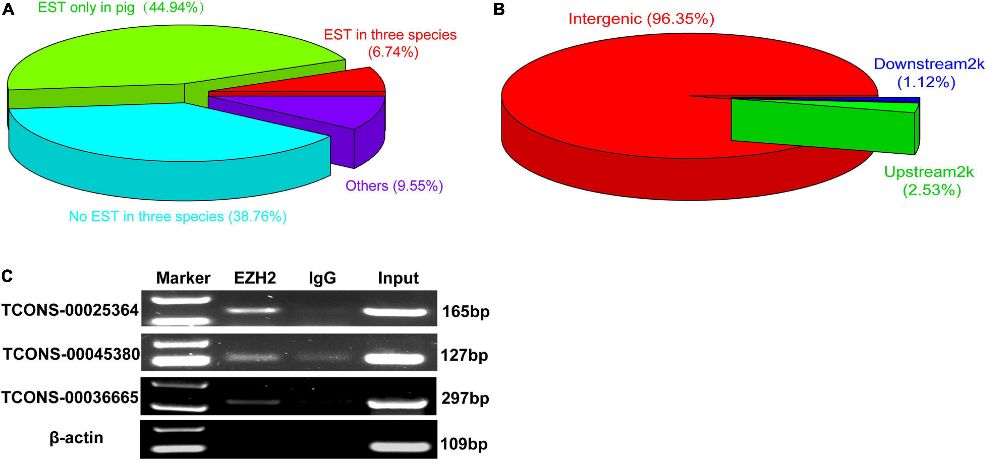 Figure 2. The verification of EZH2-binding novel lincRNAs.
Figure 2. The verification of EZH2-binding novel lincRNAs.
Three lincRNAs, TCONS-00025364, TCONS-00045380, and TCONS-00036665, were confirmed to bind EZH2 through RIP-PCR experiments (Figure 2C). TCONS-00036665, aligning with the pig EF397601 transcript, shares similarities with the human and mouse NEAT1 gene, suggesting its involvement in skeletal muscle development. Further characterization revealed TCONS-00036665 as a 3,450 bp polyadenylated transcript, predominantly localized in the nuclei of proliferating and differentiated pig muscle satellite cells (PSCs). Expression analysis indicated that TCONS-00036665 increases during PSC differentiation, suggesting its role in regulating proliferation and differentiation processes.
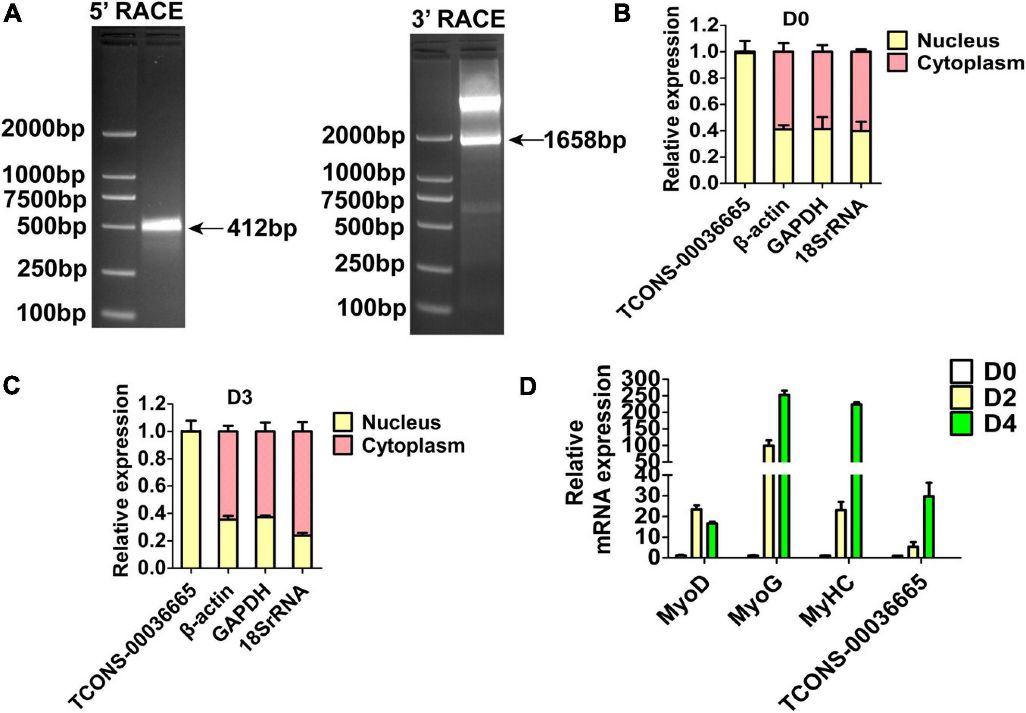 Figure 3. The molecular characterization of TCONS-00036665.
Figure 3. The molecular characterization of TCONS-00036665.
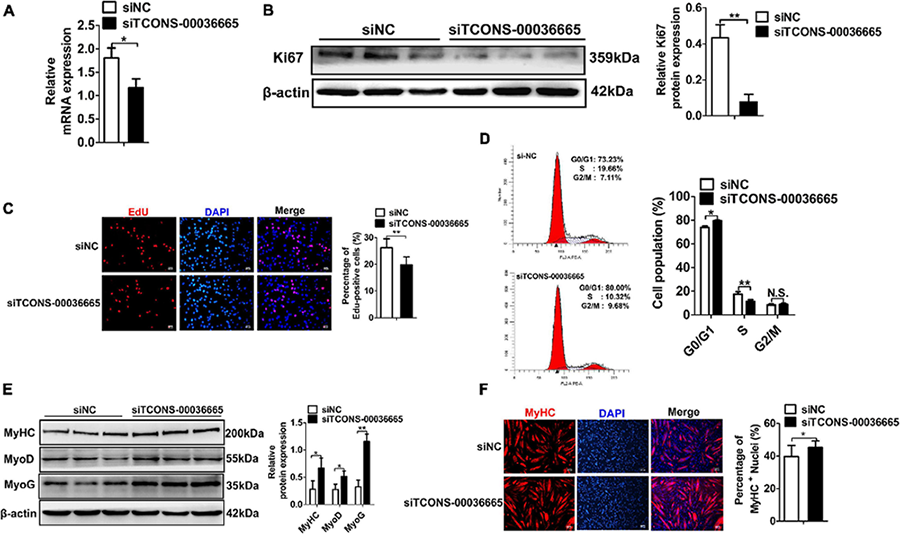 Figure 4. Knockdown of TCONS-00036665 inhibits PSC proliferation but promotes PSC differentiation.
Figure 4. Knockdown of TCONS-00036665 inhibits PSC proliferation but promotes PSC differentiation.
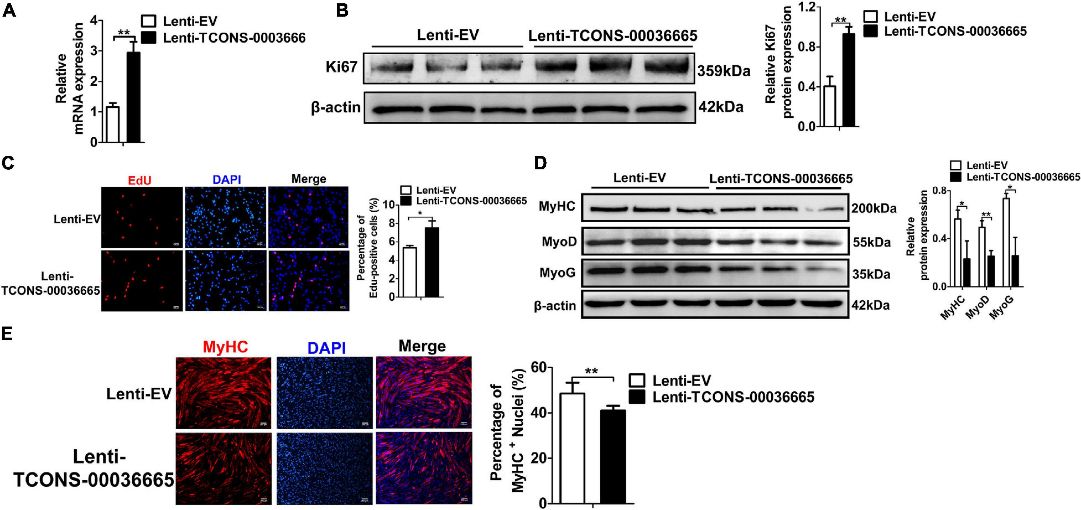 Figure 5. TCONS-00036665 overexpression promotes PSC proliferation but inhibits PSC differentiation.
Figure 5. TCONS-00036665 overexpression promotes PSC proliferation but inhibits PSC differentiation.
Conclusion
In this study, EZH2-mediated gene repression in pig skeletal muscle was investigated. TCONS-00036665, an EZH2-binding lincRNA, was identified and found to regulate muscle satellite cell proliferation and differentiation by enhancing EZH2-mediated H3K27me3 enrichment on target gene promoters. The findings suggest a novel regulatory role for lincRNAs in pig muscle development through epigenetic mechanisms involving EZH2.
Reference
- Wang S, Xu X, Liu Y, et al. RIP-Seq of EZH2 Identifies TCONS-00036665 as a Regulator of Myogenesis in Pigs. Frontiers in Cell and Developmental Biology, 2021, 8: 618617.
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


