
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics is dedicated to producing high-quality ChIP-seq data sets to help you profiling DNA targets for histone modification, structural proteins, transcription factors, and other DNA-associated proteins at a genome-wide scale.
The Introduction of ChIP-Seq
ChIP-Seq (chromatin immunoprecipitation sequencing), which refers to the binding site analysis, is a way to analyze DNA-protein interactions. The technique combines chromatin immunoprecipitation (ChIP) with NGS to identify where the DNA binds to the associated proteins. It can be used to pinpoint the binding sites of any interested protein throughout the genome. ChIP-seq is often used to determine how transcription factors and other chromatin-related proteins affect phenotypic mechanisms. Determining how proteins interact with DNA to regulate gene expression is essential to fully understand many biological processes and disease states. ChIP-Seq is a valuable tool for discerning and quantifying the specific DNA sequences where proteins bind or epigenetic modifications exist, it has played valuable roles in applications include studies on gene regulation, transcription complex assembly, DNA repair, histone modification, developmental mechanisms, disease progression and modifications.
The principle of ChIP-Seq involves the specific enrichment of DNA fragments bound by the target protein through chromatin immunoprecipitation (ChIP) technique, followed by purification and library construction. Subsequently, the enriched DNA fragments are subjected to high-throughput sequencing. Researchers meticulously map the obtained millions of sequence tags to the genome, thereby obtaining information on DNA segments interacting with histones, transcription factors, and other proteins across the entire genome.
Difference between ChIP-Seq and ATAC-Seq
ChIP-Seq and ATAC-Seq are two powerful techniques utilized in molecular biology to investigate protein-DNA interactions and chromatin accessibility, respectively.
ChIP-Seq experiments aim to validate the interaction between a specific transcription factor and DNA molecules. Prior to the experiment, researchers have already identified the transcription factor of interest. By designing antibodies targeting this transcription factor, the ChIP experiment captures DNA fragments bound to it. This method aids in identifying the interactions between specific proteins and DNA.
In contrast, ATAC-Seq technology does not target specific transcription factors but is employed to assess the open chromatin regions across the entire genome. Through this approach, potential binding sites for proteins can be identified at a genome-wide level. Integrating ATAC-Seq with other methodologies can assist researchers in identifying and characterizing regulatory elements of interest.
Advantages of ChIP-Seq
- Exceptionally high quality and high-resolution sequencing: It is possible to acquire millions of sequence tags and rare protein binding sites in the genome, ability to identify novel enrichment sites.
- Cost-effective: rapid and efficient genome-wide profiling of multiple samples in one run and only use 1/100 of the amount of DNA required for ChIP-chip.
- Comprehensive analysis: Utilizing widely accepted software and latest programs for motif prediction, peak annotation, functional analysis and data visualization of ChIP-Seq.
Applications of ChIP-Seq
- Gene regulation
- Transcription complex assembly
- DNA repair
- Histone modification
- Developmental mechanisms
- Disease progression
ChIP-Seq Workflow
A typical ChIP-Seq workflow is shown as follow:
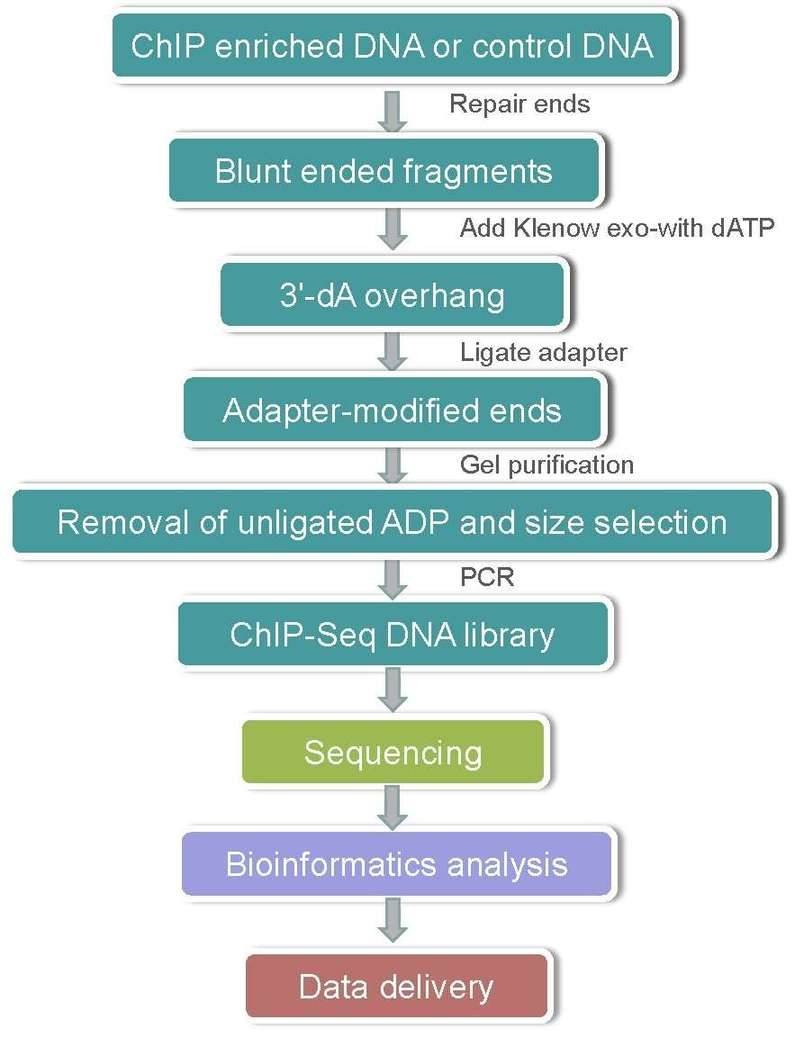
Service Specification
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Data Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
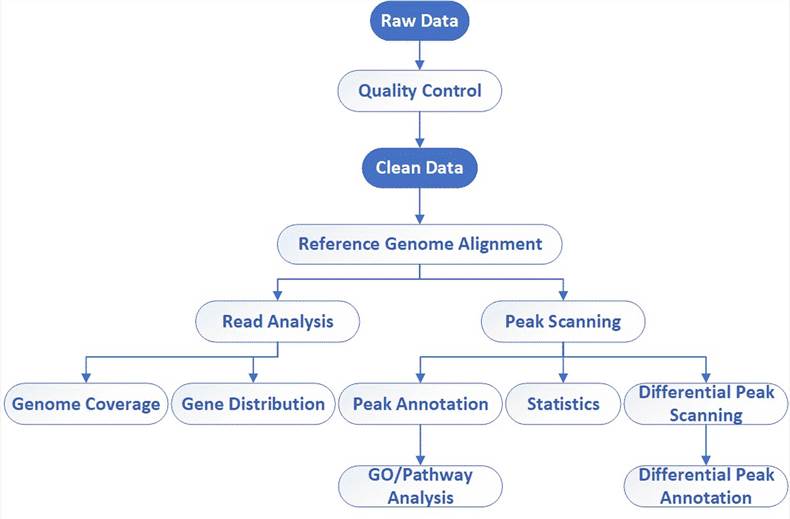
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in ChIP-Seq for your writing (customization)
At CD Genomics, we provide you with high-quality sequencing and comprehensive bioinformatics analysis for your ChIP-Seq project, enabling accurately screen and determine the protein binding sites in the whole genome. If you have additional requirements or questions, please feel free to contact us.
Reference:
- Taiwo O, Wilson G A, Morris T, et al. Methylome analysis using MeDIP-seq with low DNA concentrations. Nature protocols, 2012, 7(4): 617.
Partial results are shown below:
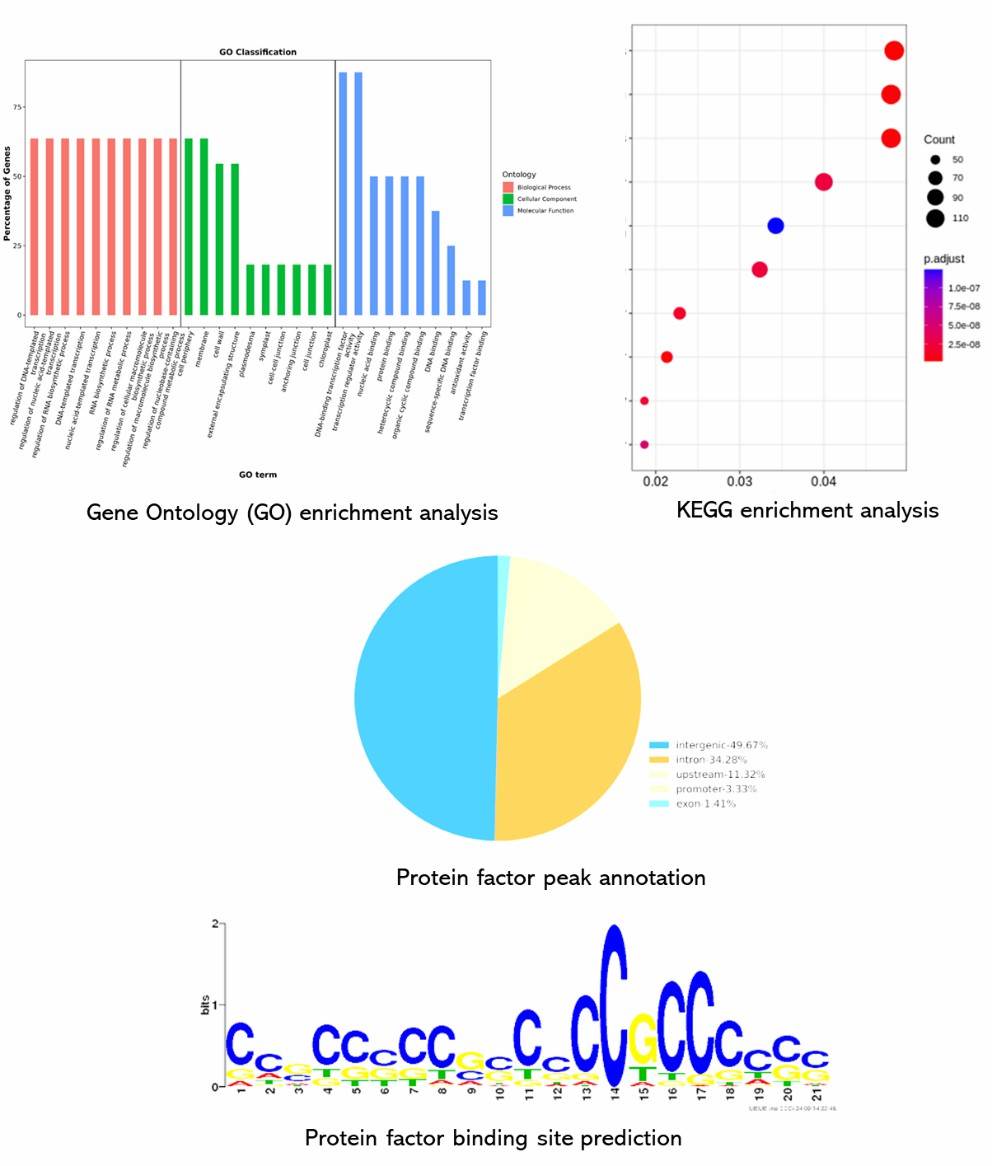
1. What are the advantages of ChIP-Seq compared to ChIP-chip?
- ChIP-Seq enables genome-wide analysis, while ChIP-chip only analyzes parts of known sequences.
- ChIP-Seq provides accurate analysis of binding sequences, while ChIP-chip could not precise positioning.
2. What is the difference between sonication- and enzymatic-based chromatin fragmentation?
Either sonication or enzymatic digestion can be used to generate the appropriate lengths of chromatin. Sonication is a traditional method used for fragmenting chromatin, it uses acoustic energy to forcefully shear the chromatin. Sonicated chromatin works very well for performing ChIP to assess histones and histone modifications, however, over-sonication can damage the chromatin and displace bound transcription factors and cofactors. Therefore, sonication typically needs optimization. Enzymatic digestion uses micrococcal nuclease to cut the linker region between nucleosomes. It gently fragments the chromatin and preserves the integrity of chromatin and bound proteins. So it's more suitable for performing ChIP to assess transcription factors and cofactors that are less abundant and interact with DNA less stable. In addition, enzymatic digestion provides better reproducibility between experiments. But over-digestion may lead to a loss of nucleosome-free regions.
3. Will high-resolution be guaranteed as long as high-throughput sequencing is used?
High throughput sequencing has greatly improved the resolution of ChIP detection, but it is not the only determinant of high resolution. After immunoenrichment, the length of fragments interrupted by chromatin also affects resolution.
4. What are the factors that lead to a higher false positive for ChIP-seq?
DNA fragmentation methods, heterogeneity of chromatin openness, PCR amplification bias, genome duplication, and errors in sequencing and sequence alignments lead to false positive. After sequencing, the sequences are compared to the known genome and identified the true binding sites (peaks). For transcription factors, look for the downstream regulatory gene (target gene) corresponding to the "peak" or construct a conserved binding sequence for the transcription factor binding site. If the motif of the transcription factor is known, calculate the percentage of the motif sequence in the "peak" sequence, and the reliability of the experimental results can be estimated.
ChIP-seq analysis of histone H3K9 trimethylation in peripheral blood mononuclear cells of membranous nephropathy patients
Journal: Brazilian Journal of Medical and Biological Research
Impact factor: 2.904
Published: 12 December 2013
Background
Membranous nephropathy (MN), characterized by the presence of diffuse thickening of the glomerular basement membrane and subepithelial in situ immune complex disposition, is the most common cause of idiopathic nephrotic syndrome in adults, with an incidence of 5-10 per million per year. A number of studies have confirmed the relevance of several experimental insights to the pathogenesis of human MN, but the specific biomarkers of MN have not been fully elucidated. High-throughput techniques, such as Chromatin Immunoprecipitation followed by sequencing (ChIP-seq), provide a thorough examination of gene expression and disease processes at the genomic scale. In this investigation, the profiling of H3K9me3 alterations in peripheral blood mononuclear cells (PBMCs) sourced from individuals with minimal change nephrotic syndrome (MN) and unaffected individuals serves to illuminate the pathogenic pathways implicated in MN.
Materials & Methods
- 10 MN patients and 10 healthy volunteers
- Blood samples
- Isolation of PBMCs
- Chromatin immunoprecipitation
- Next-generation sequencing
- Data processing and alignment
- Peak identification
- Gene annotation
Results
In this study, the authors used chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) to analyze the variations in a methylated histone (H3K9me3) in peripheral blood mononuclear cells from 10 MN patients and 10 healthy subjects. There were 108 genes with significantly different expression in the MN patients compared with the normal controls. In MN patients, significantly increased activity was seen in 75 H3K9me3 genes, and decreased activity was seen in 33, compared with healthy subjects. Five positive genes, DiGeorge syndrome critical region gene 6 (DGCR6), sorting nexin 16 (SNX16), contactin 4 (CNTN4), baculoviral IAP repeat containing 3 (BIRC3), and baculoviral IAP repeat containing 2 (BIRC2), were selected and quantified. There were alterations of H3K9me3 in MN patients.
Table.1 ChIP-seq and alignment results.
 Aligned reads: the number of reads refers to the reference genome; Aligned reads only: the number of reads only refers to the reference genome; % Aligned: aligned reads/total reads; % Only: aligned reads only/total reads.
Aligned reads: the number of reads refers to the reference genome; Aligned reads only: the number of reads only refers to the reference genome; % Aligned: aligned reads/total reads; % Only: aligned reads only/total reads.
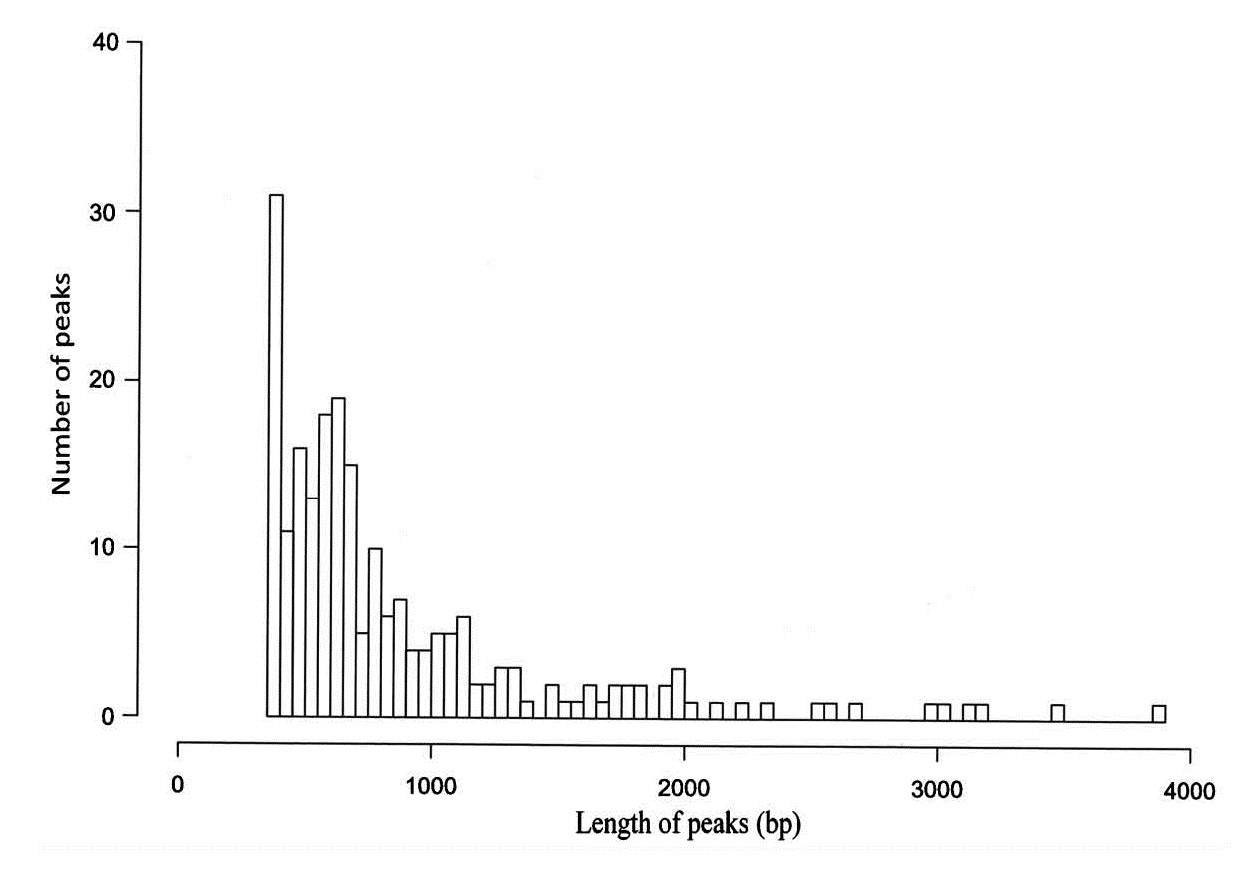 Figure 1. ChIP-seq peak distribution (distance) of H3K9me3 from membranous nephropathy patients.
Figure 1. ChIP-seq peak distribution (distance) of H3K9me3 from membranous nephropathy patients.
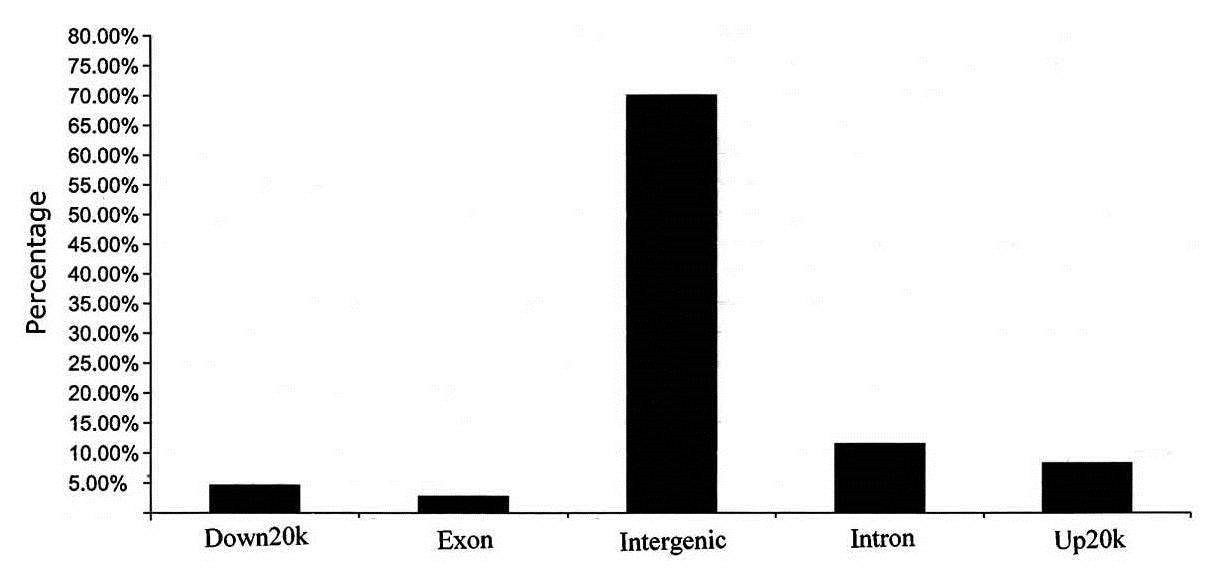 Figure 2. Genome-wide distribution of peaks relative to annotated genes.
Figure 2. Genome-wide distribution of peaks relative to annotated genes.
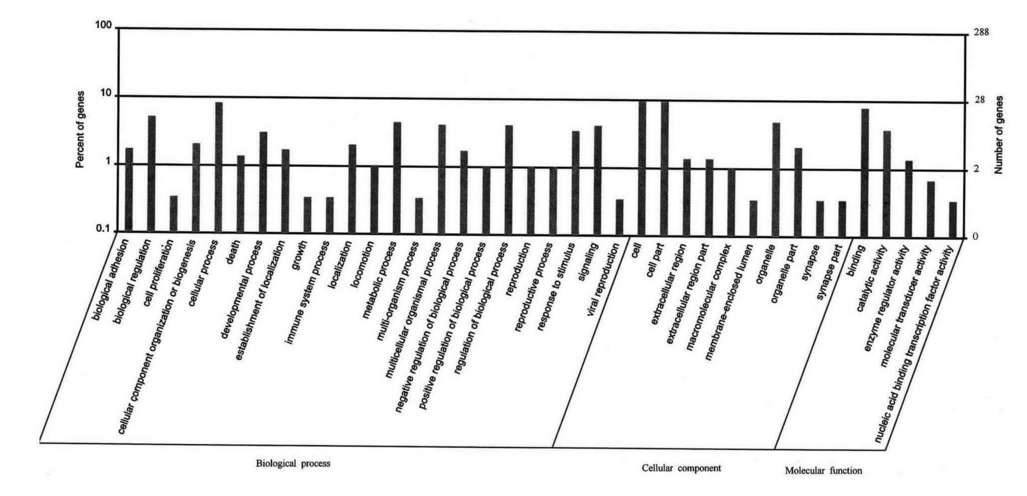 Figure 3. Distribution of peaks relative genes by Gene Ontology (GO) analysis (AmiGO 1.8). The lateral axis represents the GO terminology. The left vertical axis represents the proportion of the related genes. The right vertical axis represents the number of the related genes.
Figure 3. Distribution of peaks relative genes by Gene Ontology (GO) analysis (AmiGO 1.8). The lateral axis represents the GO terminology. The left vertical axis represents the proportion of the related genes. The right vertical axis represents the number of the related genes.
Table 2. Basic information of motif of H3K9me3 from patients with membranous nephropathy.
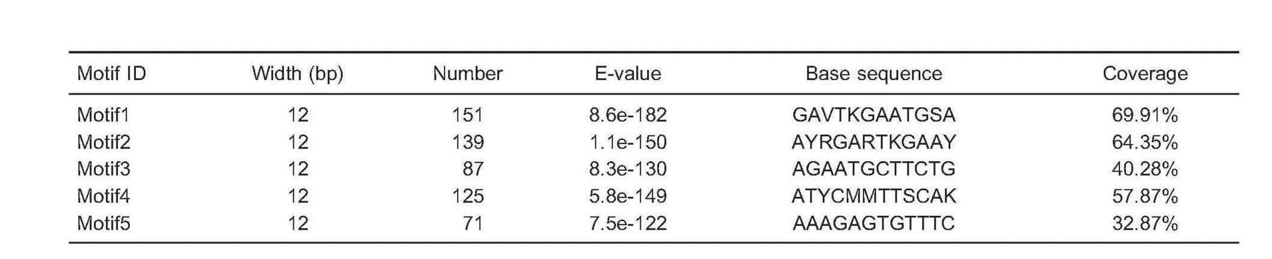 S: C+G; R: A+G; K: T+G; Y: C+T; M: A+C; V: A+C+G.
S: C+G; R: A+G; K: T+G; Y: C+T; M: A+C; V: A+C+G.
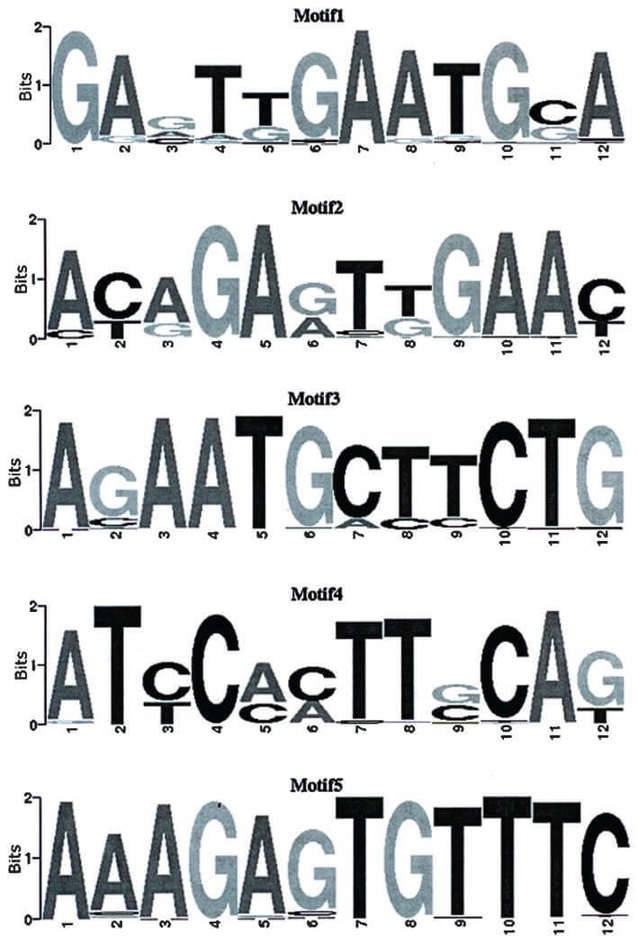 Figure 4. ChIP-seq motif logo. The lateral axis indicates locus of motif. The total height of the vertical axis reflects the conservation of the motif. The height of each base represents probability of the base.
Figure 4. ChIP-seq motif logo. The lateral axis indicates locus of motif. The total height of the vertical axis reflects the conservation of the motif. The height of each base represents probability of the base.
Table 3. Selected genes with H3K9me3 alterations between patients with membranous nephropathy and healthy controls identified by ChIP-seq.
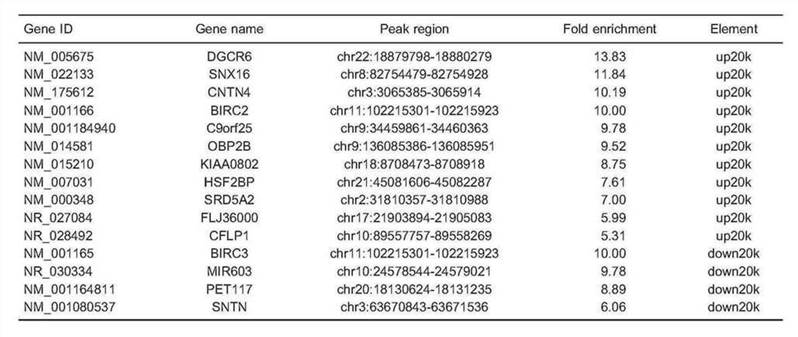
Conclusion
In summary, the authors think these results may be candidates to help explain pathogenesis in MN patients. Such novel findings show that H3K9me3 may be a potential biomarker or promising target for epigenetic-based MN therapies.
Reference:
- Sui W G, et al. ChIP-seq analysis of histone H3K9 trimethylation inperipheral blood mononuclear cells of membranous nephropathy patients. Brazilian journal of medical and biological research, 2014, 47(1):42-9.
Here are some publications that have been successfully published using our services or other related services:
Downregulated PITX1 Modulated by MiR-19a-3p Promotes Cell Malignancy and Predicts a Poor Prognosis of Gastric Cancer by Affecting Transcriptionally Activated PDCD5
Journal: Cellular Physiology and Biochemistry
Year: 2018
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


