
 Sample Submission Guidelines
Sample Submission Guidelines
With decades of experience in the fields of genomics sequencing, CD Genomics is devoted to providing unprecedented amounts of microbial metatranscriptomic data. Our strong expertise in the informative and unbiased metatranscriptomic sequencing service is guaranteed by state-of-the-art high throughput sequencers, flexible sequencing strategies, and professional bioinformatics pipelines.
The Introduction of Metatranscriptomic Sequencing
Metatranscriptomics is the culture-independent profiling (including protein-coding and non-coding DNA) of microbial community-wide gene expression, which is capable of monitoring RNA-based regulation and expressed biological signatures of complex bacterial communities in a given sample at a given moment and under specific conditions. It elucidates three aspects of a microbial community, including gene activity diversity, gene expression abundance, and differential gene expression analysis. The gene expression analysis can tell which genes exhibit the highest change in expression levels in different conditions potentially to identify biomarkers and expression signatures.
While metagenomics shows us the microbial species present in the community and their genomic potentials, metatranscriptomics presents the function and activity of the complete set of transcripts, as well as community structure. Metatranscriptomics offers a more informative perspective compared with metagenomics in revealing active biochemical functions, which has become a focus for applications in the environmental, medical, and energy fields as well as the field of drug discovery.
Advantages of Metatranscriptomic Sequencing
- A culture-free method to reveal the true extent of microbial diversity
- Permitting function-based activity screens
- More targeted than shotgun random sequencing
- Cost-efficient and time-effective
- A wide range of applications, including basic research, ecological applications, clinical applications, industrial applications, and so on
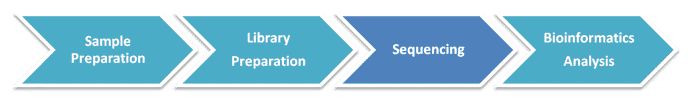
Metatranscriptomic Sequencing Workflow
Our highly experienced expert team executes quality management following every procedure to ensure confident and unbiased results. The general workflow for metatranscriptomic sequencing is outlined below. After RNA isolation from qualified samples, rRNA depletion, library construction, high-throughput sequencing, and bioinformatics analysis are in turn performed. We can help you make decisions on the library construction method, the depth of coverage, and the data analysis strategies based on your research aims.
Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
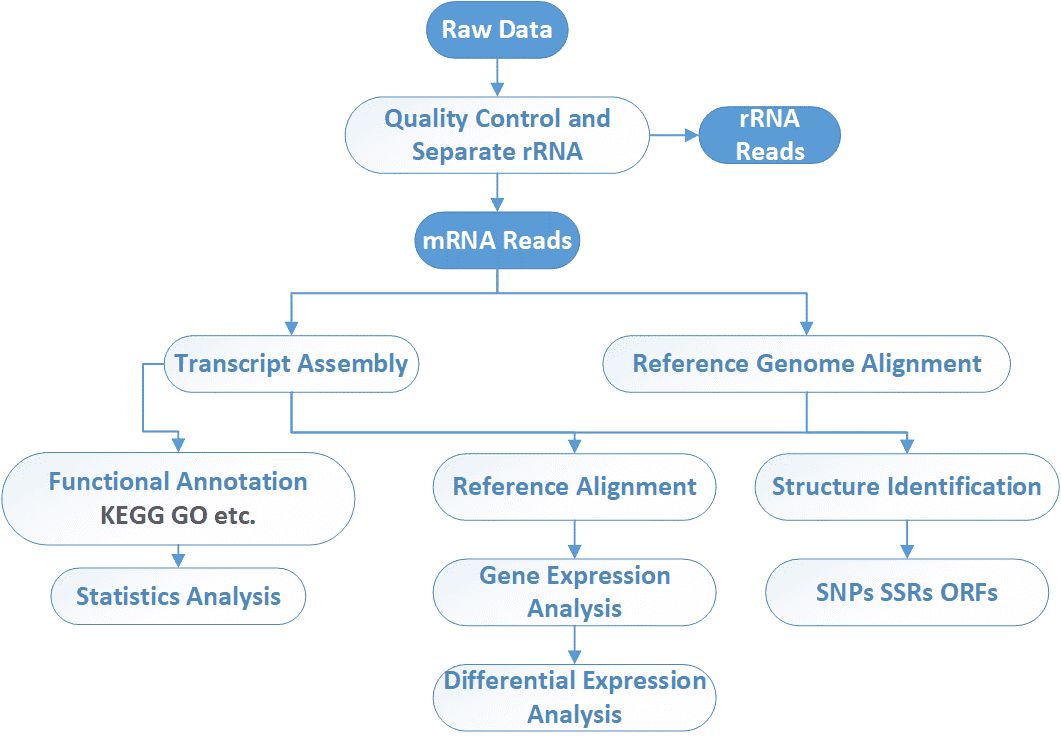
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Metatranscriptomic Sequencing for your writing (customization)
CD Genomics provides full metatranscriptomic sequencing service package including sample standardization, library construction, Hiseq sequencing, raw data alignment, down-stream bioinformatics processing and statistical analysis. We can tailor this pipeline to your research interest. If you have additional requirements or questions, please feel free to contact us, our specialists are more than happy to assist you.
Reference
- Warnecke F, Hess M. A perspective: metatranscriptomics as a tool for the discovery of novel biocatalysts. Journal of Biotechnology, 2009, 142(1): 91-95.
Partial results are shown below:
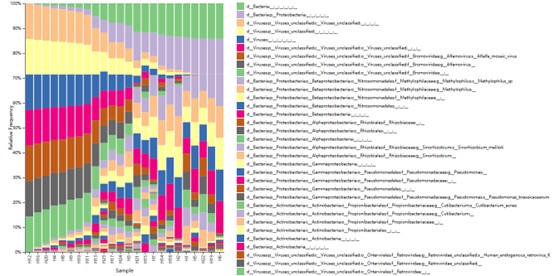
The taxonomy distribution of all sample in Phylum classification level.
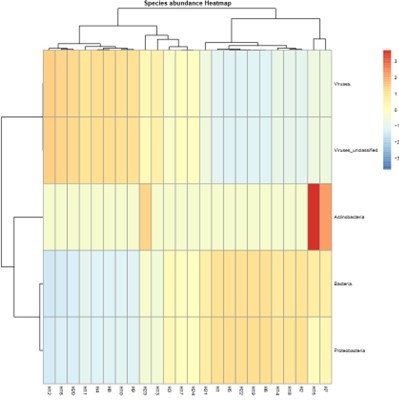
Species abundance Heatmap.
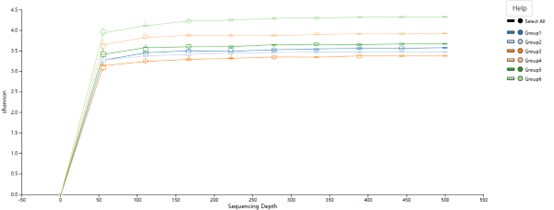
Rarefaction curve of the sequenced reads for all samples.
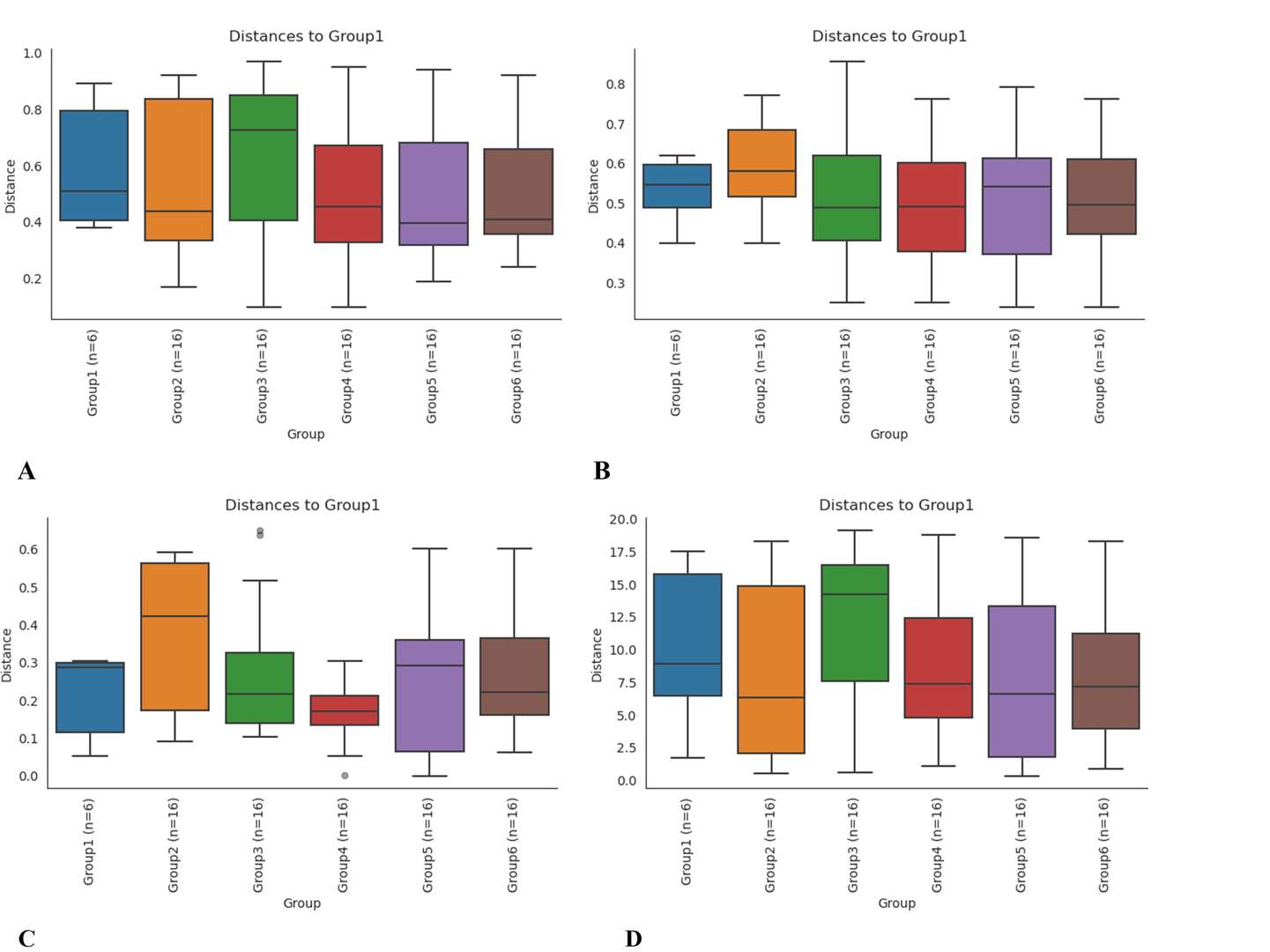
Boxplot analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).
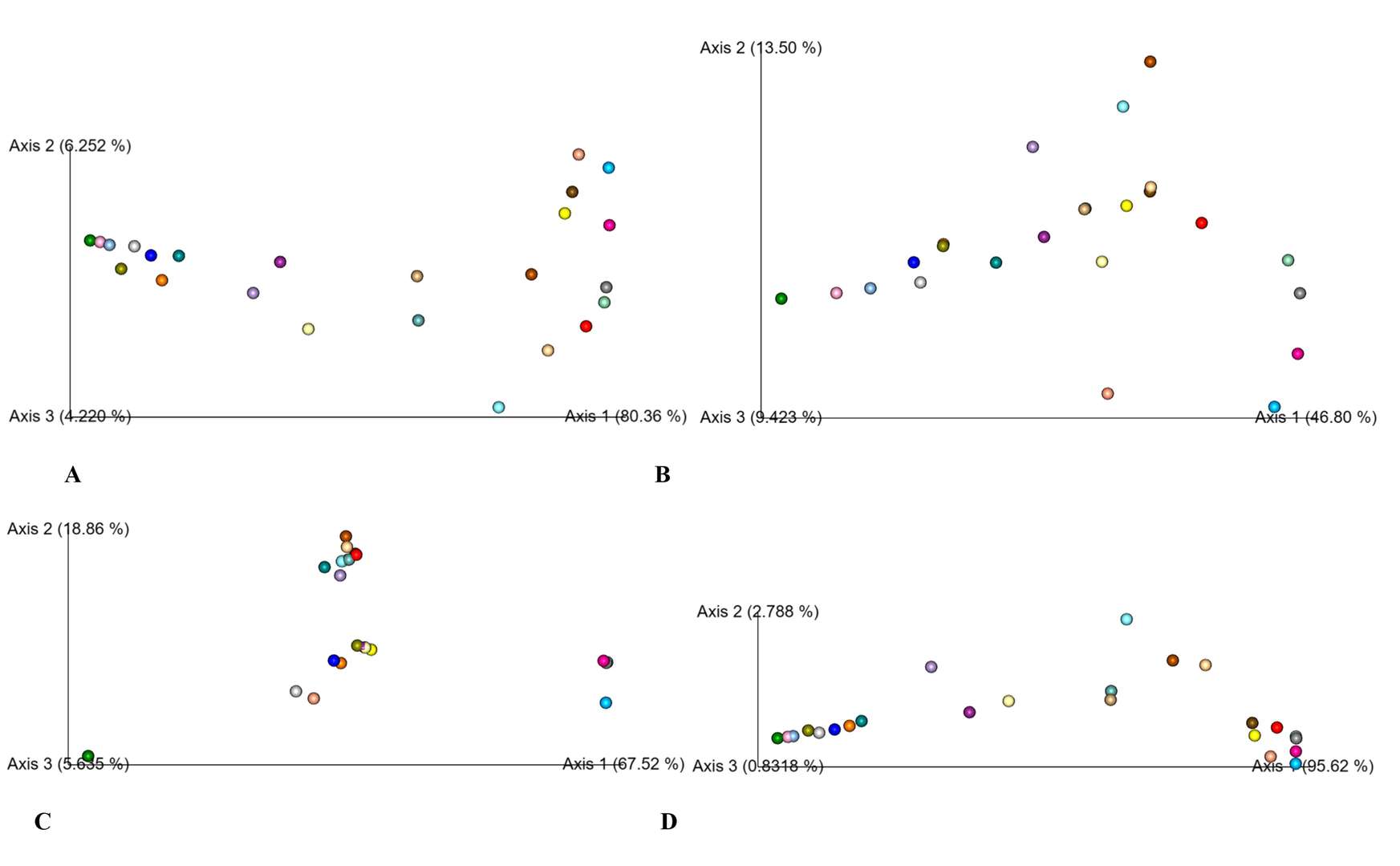
PCoA analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).
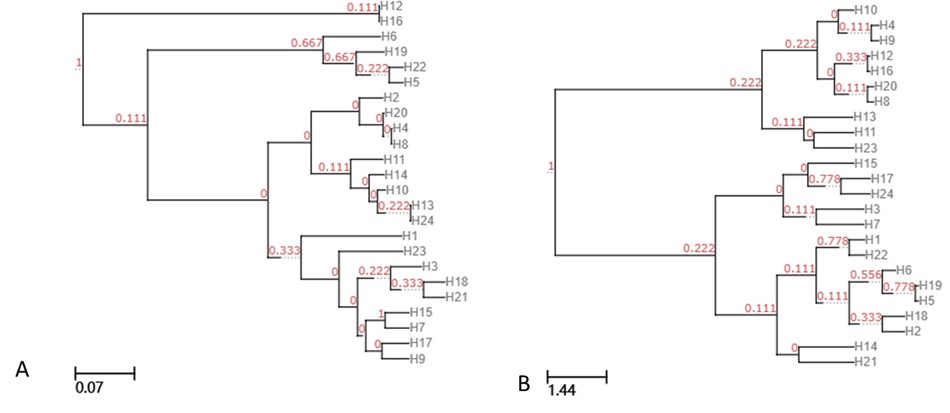
UPGMA clustering tree based on unweighted unifrac (A), and weighted unifrac (B).
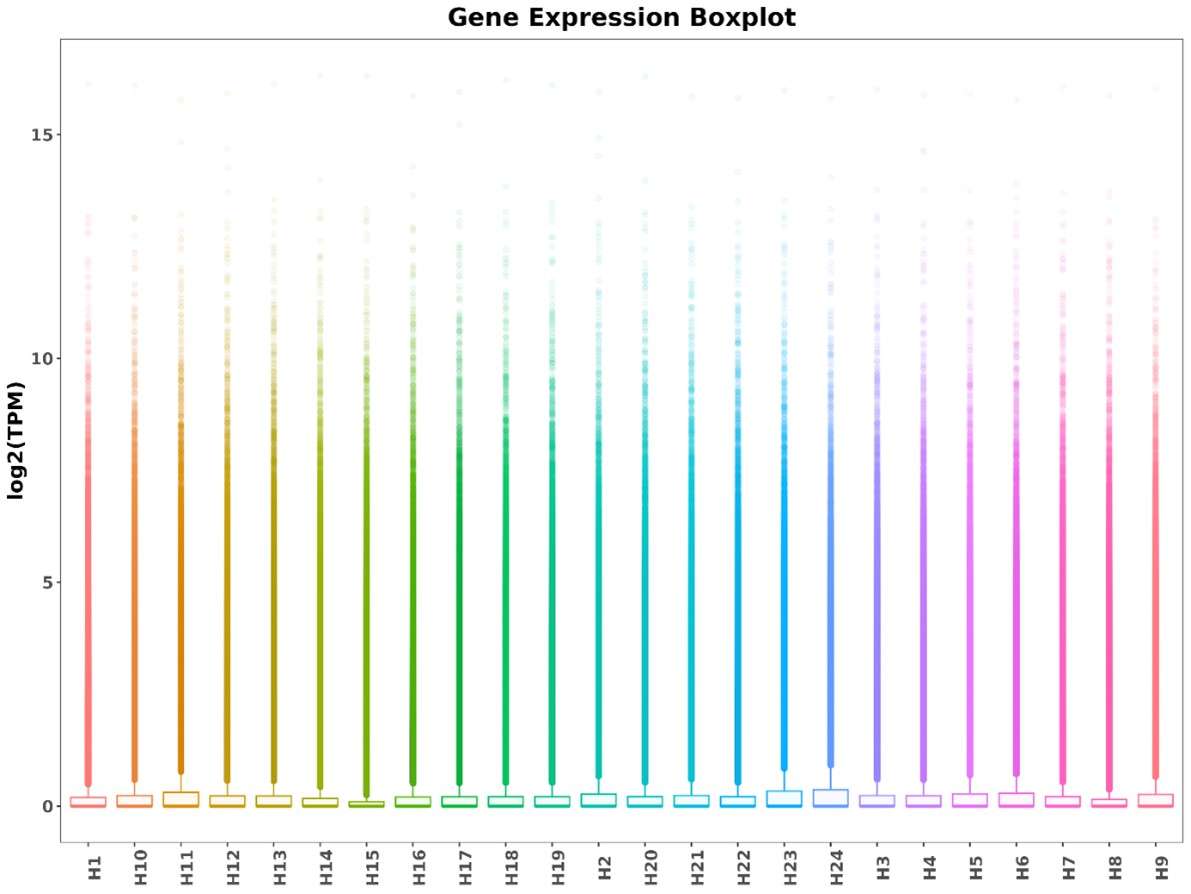
Boxplot of TPM for each sample.
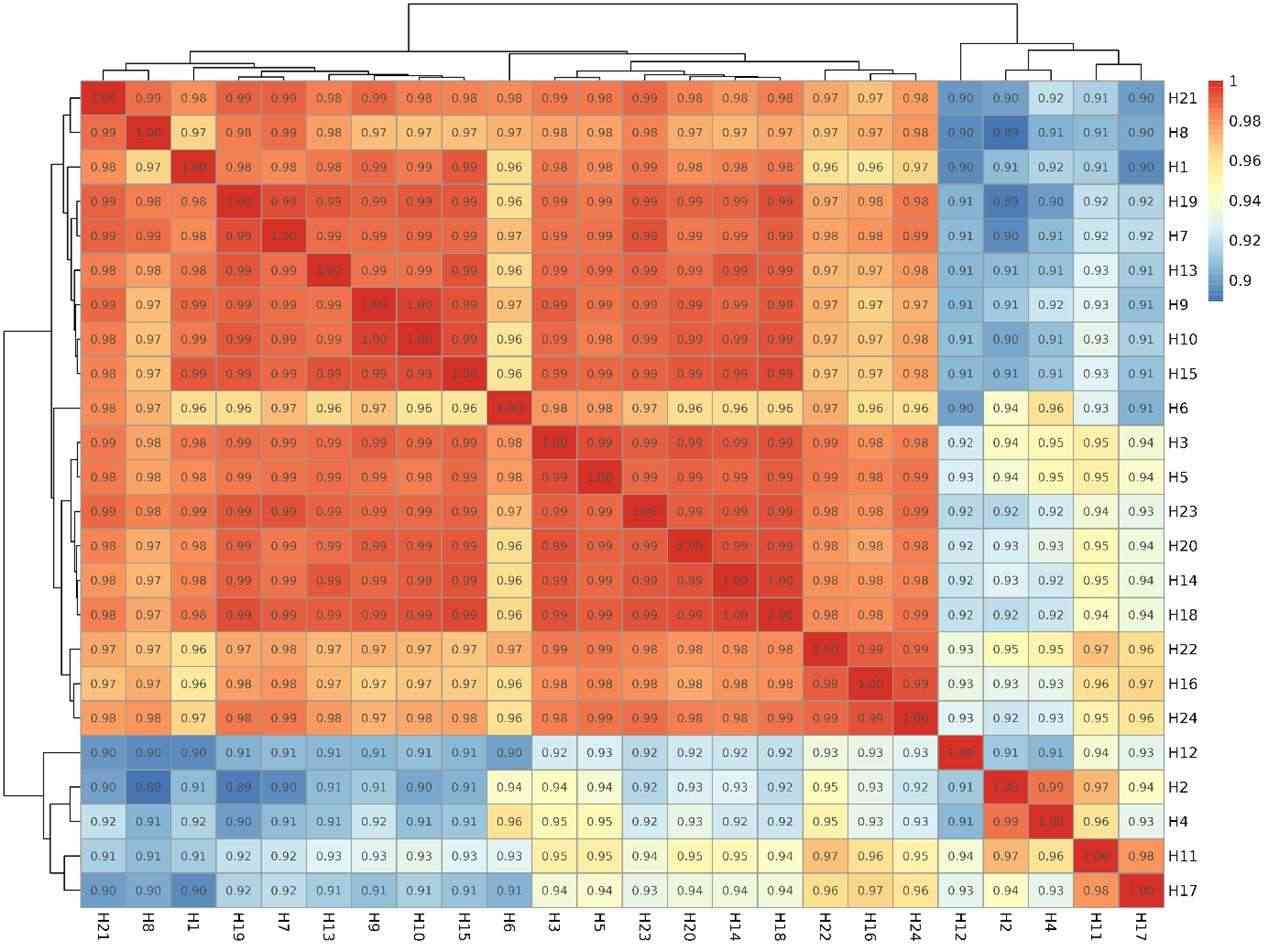
Correlation graph of gene number.
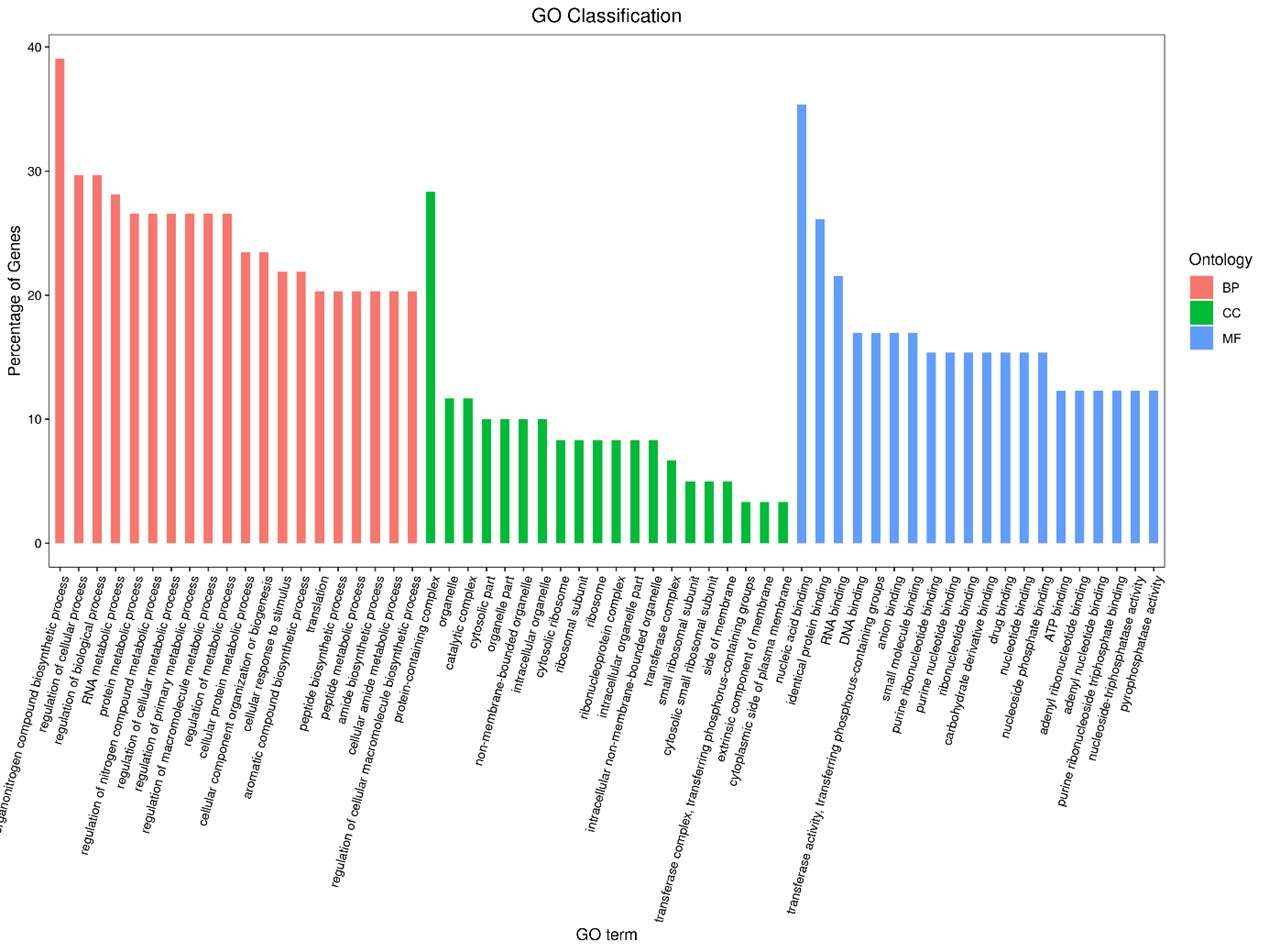
Statistics results of GO annotation for CLC_vs_SLC.
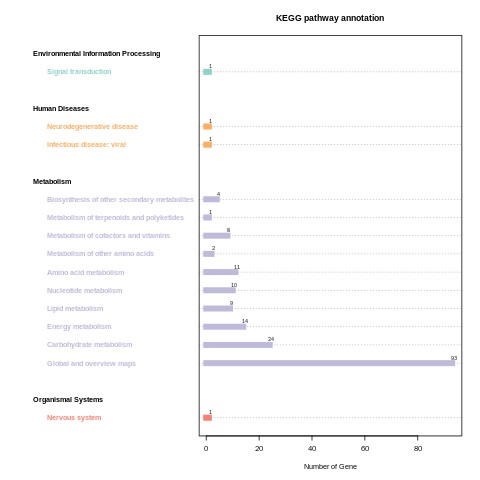
CLC_vs_SLC KEGG_classification.

Statistical of specific function database common and unique annotation.

CAZy function classification.
1. What are the noteworthy issues of RNA samples?
The contamination should be rigorously excluded when sampling. In detail, sampling-related instruments and consumables should be sterilized and RNase-free. The freshly obtained samples should be immediately frozen by putting into liquid nitrogen, or directly submitting original environmental or clinical samples to us. The recommended total RNA amount for submission is 6 µg or more with a concentration of greater than 50 ng/µl.
2. What kind of QC methods do you adopt for the customer's samples?
We will perform QC on your total RNA samples prior to sequencing them. We use the Agilent Bioanalyzer to determine the RNA Integrity Number (RIN). If the RIN is lower than 8, the samples will not pass QC. The library QC will also be performed using the Agilent Bioanalyzer to determine library size and purity. Also, prior to loading the libraries on the sequencer, we perform qPCR quantification. The cost for this is included in the sequencing service. The raw data will pass our Q30 filter, which means more than 80% of bases with a greater than Q30 quality score.
3. What are the advantages of metatranscriptomics?
Metatranscriptomics is the genomic analysis of complete microbial transcriptomes, providing a particularly rich source of data on the global diversity of RNA viruses and their evolutionary history. Metatranscriptomics has several advantages over traditional methods such as cell culture, consensus PCR, and metagenomics approaches based on viral particle purification.
Metatranscriptomics has proven successful in characterizing the RNA viromes of diverse invertebrates. Specifically: (i) it uncovers the entire RNA virome, with sufficient coverage to assembly complete viral genomes, including those from co-infecting parasites; (ii) it offers a reliable quantification and assessment of both viral and host RNAs; (iii) it is comparatively simple, requiring minimal sample processing; and (iv) it provides more information than the genome sequence alone, allowing a characterization of viral diversity and ecology.
References
- Shi M, Neville P, Nicholson J, et al. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. Journal of Virology, 2017, 91(17): e00680-17.
- Shi M, Zhang Y Z, Holmes E C. Meta-transcriptomics and The Evolutionary Biology of RNA Viruses. Virus research, https://doi.org/10.1016/j.virusres.2017.10.01
High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia
Journal: Journal of Virology
Impact factor: 4.663
Published: 21 June 2017
Background
Mosquitoes harbor a high diversity of RNA viruses, including many that impact human health. Although many efforts to reveal the extent and nature of the mosquito virome, little is known about how these viruses persist, spread, and interact with both their hosts and other microbes. To infer features of virome ecology among mosquito species from different geographic locations, the authors characterized the total transcriptome of 12 populations.
Methods
Sample collection
A total of 519 adult mosquitoes collected from four locations in Western Australia
Species identification
Taxonomic keys
Dissecting microscopes
cox1 gene
Sequencing
RNA isolation and quility control
Sequencing: HiSeq 2500, paired-end (100 bp)
RNA virus discovery and genome anonotation
RNA quantification
Phylogenetic analyses
Results
1. The mosquito virome
Blast analyses showed the complete genomes of 24 RNA viruses species, of which 19 were newly described here. For each library, the number of virus species varied from 1 to 10 (Table 1).
![]()
2. Virome ecology
The virus composition and abundance revealed substantial differences between the Culex and Aedes genera (Figure 1). Generally, the Aedes mosquitoes harbour fewer viruses. Of the 24 viral species discovered, only Wilkie qin-like virus (WQLV) and Wilkie narna-like virus 1 (WNLV1) were shared between the two genera (Figure 2).
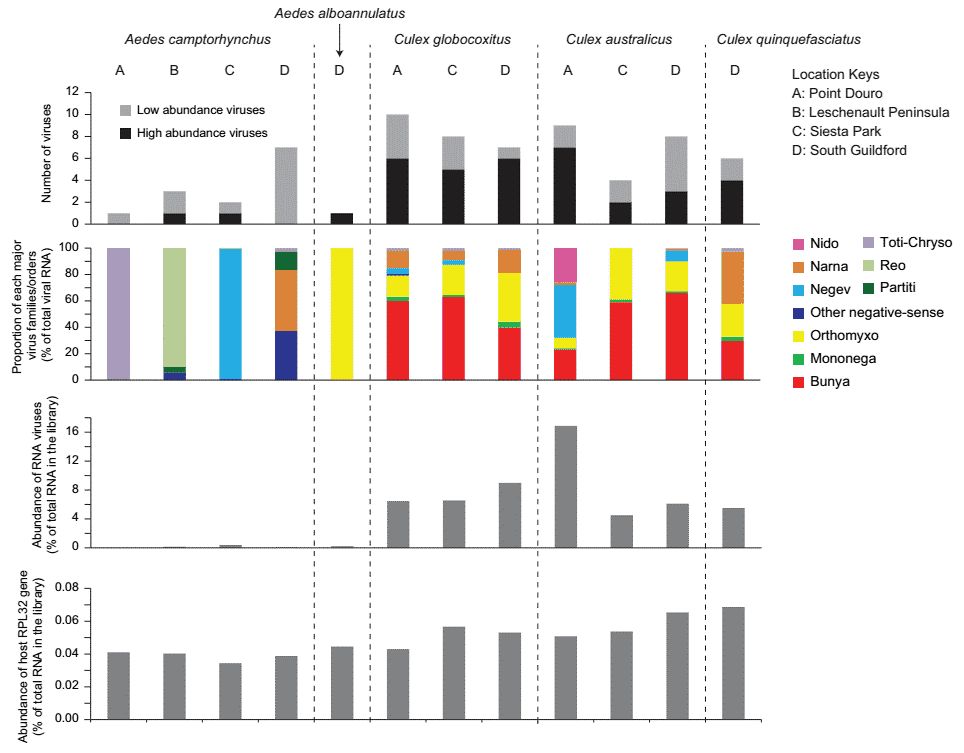 Figure 1. An overview of the diversity and abundance of the RNA viruses discovered.
Figure 1. An overview of the diversity and abundance of the RNA viruses discovered.
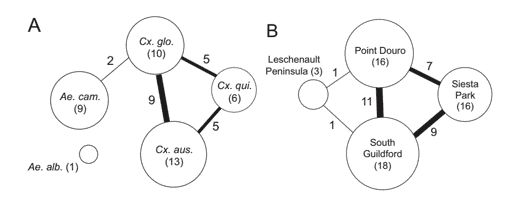 Figure 2. The similarity of viromes between host species (A) and geographic locations (B).
Figure 2. The similarity of viromes between host species (A) and geographic locations (B).
3. Evolutionary analysis
The authors discovered eight putative negative-sense RNA viruses and seven double-stranded RNA viruses. The positive-sense RNA viruses discovered fell with the Narnaviridae, Mesoniviridae, Negev-like viruses, and Luteoviridae-related viruses. The authors performed phylogenetic analyses and genomic characterizations (please refer to the original paper for more detailed phylogenetic information). interestingly, two viruses (WQLV and WPLV2) were co-appeared with a group of fungi termed "Unknown sp1, 2, and 3" and they had matching evolutionary histories (Figure 3).
 Figure 3. The matching tree topologies of the Wilkie qin-like viruses and a group of fungi (cox 1 gene) discovered in three mosquito pools.
Figure 3. The matching tree topologies of the Wilkie qin-like viruses and a group of fungi (cox 1 gene) discovered in three mosquito pools.
Reference
- Shi M, Neville P, Nicholson J, et al. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. Journal of Virology, 2017, 91(17): e00680-17.
Here are some publications that have been successfully published using our services or other related services:
Transferrable protection by gut microbes against STING-associated lung disease
Journal: Cell Reports
Year: 2021
Microbial adaptation and response to high ammonia concentrations and precipitates during anaerobic digestion under psychrophilic and mesophilic conditions
Journal: Water Research
Year: 2021
Algal-bacterial synergy in treatment of winery wastewater
Journal: NPJ Clean Water
Year: 2018
Blocking IL-17A prevents oxycodone-induced depression-like effects and elevation of IL-6 levels in the ventral tegmental area and reduces oxycodone-derived physical dependence in rats
Journal: Brain, Behavior, and Immunity
Year: 2024
Indole-3-Propionic Acid, a Gut Microbiota Metabolite, Protects Against the Development of Postoperative Delirium
Journal: Annals of Surgery
Year: 2023
Elucidating the effects of organic vs. conventional cropping practice and rhizobia inoculation on rhizosphere microbial diversity and yield of peanut
Journal: Environmental Microbiome
Year: 2023
See more articles published by our clients.


