
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics offers the transcriptome-wide analysis of the location of m6A in mRNA transcripts and other RNAs by methylated RNA immunoprecipitation sequencing (MeRIP-seq). MeRIP-seq uses an antibody that specifically detects and characterizes of N6-methyladenosine (m6A) in RNA. As 3'UTR of mRNA plays important role in mRNA stability, localization, and translation, and may affect the binding of RNA-regulatory proteins to these regions, enrichment of m6A in this region indicates that m6A may affect RNA metabolism and gene expression. Inhibition of the enzymes that involve in the RNA methylation has also provided clues to the potential biological roles of this modification. Hence, the identification and characterization of modified bases in RNA may lead to a better understanding of biological pathways.
What Is MeRIP sequencing
N6-methyladenosine (m6A), the most prevalent form of methylation modification within the mRNA sequences of eukaryotes, has the capacity to functionally regulate the eukaryotic transcriptome, thus influencing the processes of mRNA splicing, nuclear export, localization, translation, and stability. The presence of m6A RNA in various vital cellular life processes signifies that mRNA methylation participates in numerous biological processes, such as stem cell differentiation and circadian rhythm, and is implicated in the pathogenesis of diverse diseases, including but not limited to carcinogenesis, obesity and infertility.
The primary technique for transcriptome research into N6-methyladenosine (m6A) is MeRIP-seq (methylated RNA immunoprecipitation sequencing). The principle underpinning this method lies in the use of m6A-specific antibodies to immunoprecipitate RNA fragments with m6A modifications within the cell, followed by high-throughput sequencing of these enriched RNA fragments. Coupled with bioinformatics analysis, thorough systematic research into m6A modifications can be performed at a transcriptome-wide level.
Advantages of Our MeRIP Sequencing Service
- High flexibility: Capable of directly sequencing high methylated fragments of the transcriptome from any species without the need for known genome sequence information.
- High precision: Able to precisely locate within a range of 100-200 nucleotides around the actual binding site.
- Digital signal: Enables quantitative and sequencing analysis directly on methylated fragments, eliminating cross-reactivity and background noise issues associated with traditional chip hybridization fluorescent analog signals.
- Comprehensive platform: MeRIP experiment—Library construction sequencing analysis—MeRIP qPCR validation, providing truly one-stop service.
- Flexible selection: Options for library construction methods and fragment sizes.
- Good reproducibility: Abundant project experience ensures experimental reproducibility, with comparative analysis conducted among multiple samples.
Application of MeRIP Sequencing
- Distribution of RNA methylation in eukaryotic organisms;
- mRNA methylation in animals and plants and its response mechanism to environmental stress;
- Mechanisms of mRNA degradation resistance;
- Mechanisms underlying tumor initiation and proliferation;
- Biological function studies of different plants;
- Cancer initiation and metastasis; Disease onset and progression;
- Cell differentiation, embryonic development, and stress response.
MeRIP Sequencing Workflow
CD Genomics performs the m6A mapping approach using immunoprecipitation with m6A specific antibodies, followed by NGS (MeRIP-Seq). Poly(A)-selected RNA was fragmented into 100-nucleotide-long oligonucleotides (input) and immunoprecipitated using an anti-m6A affinity purified antibody and then subjected to Illumina next-generation sequencing. Because immunoprecipitated RNA fragments can contain m6A anywhere along their length, multiple different m6A-containing fragments generate overlapping reads. m6A sites were bioinformatically predicted by using a peak-detection algorithm and by searching for the presence of a subset of DRACH motifs near the point of highest read coverage.
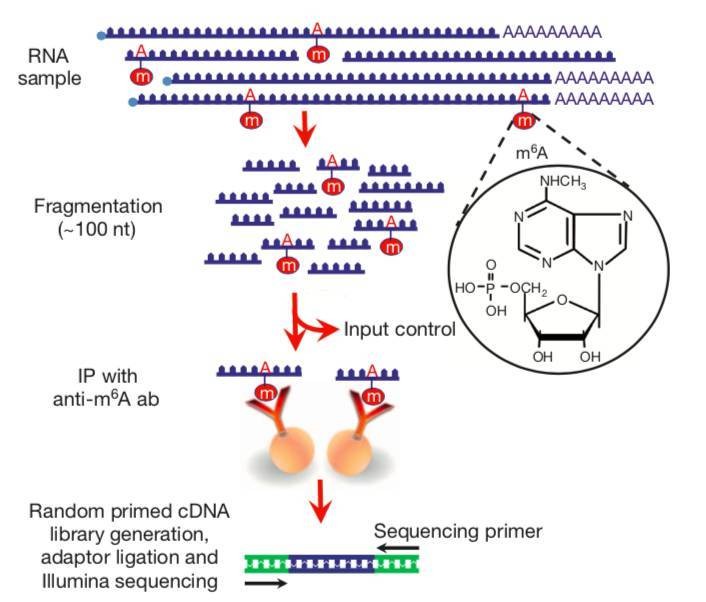 Fig. 1 MeRIP sequencing protocol
Fig. 1 MeRIP sequencing protocol
Service Specifications
Sample requirements
|
|
 |
Sequencing
|
 |
Bioinformatics Analysis
|
Analysis Pipeline
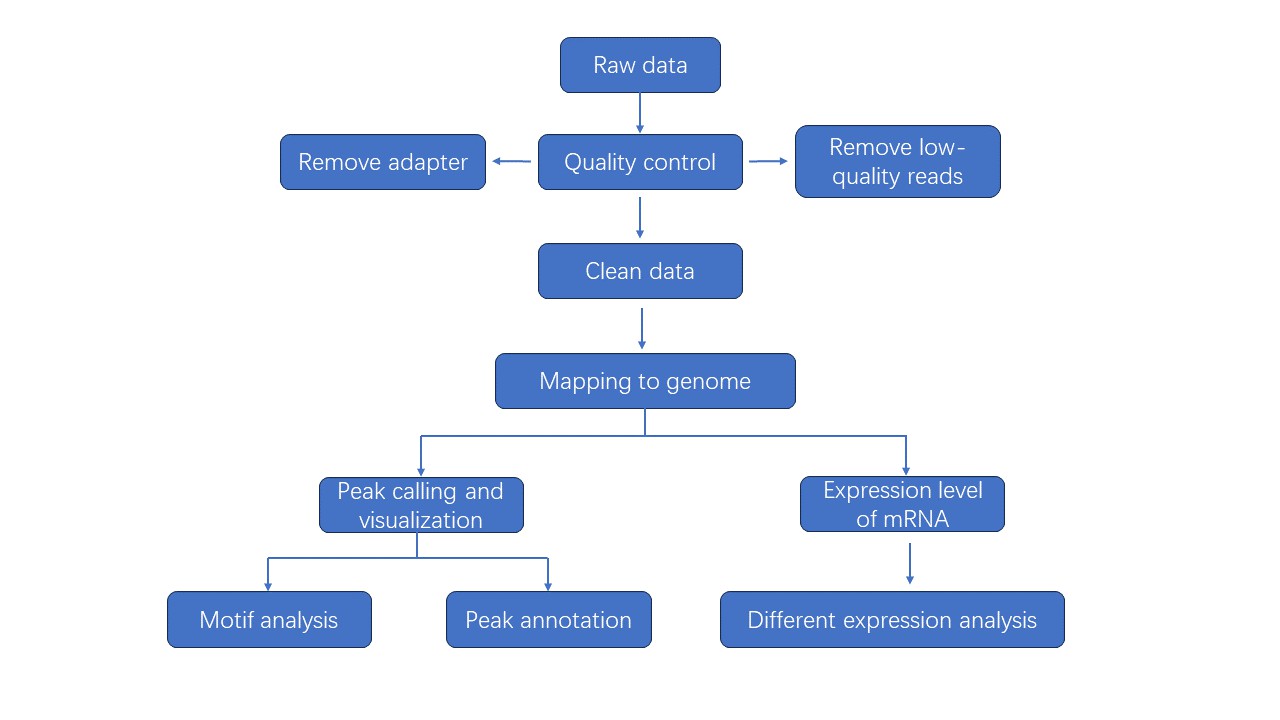
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in MeRIP sequencing for your writing (customization)
CD Genomics's MeRIP sequencing conference focuses on the links between cell variation in tissues and organ function and further elucidates the origins of diseases. If you have additional requirements or questions, please feel free to contact us.
References:
- Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 2012 Apr 29; 485(7397):201-6.
- Jesus D F D, et al. m6A mRNA Methylation Regulates Human β-Cell Biology in Physiological States and in Type 2 Diabetes. Nature Metabolism. 2019,1, 765–774.
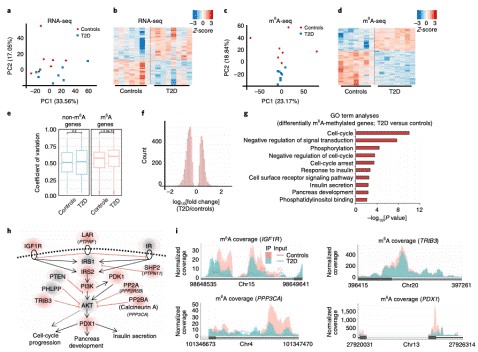 m6A mRNA Methylation Regulates Human β-Cell Biology in Physiological States and in Type 2 Diabetes. (Jesus et al., 2019)
m6A mRNA Methylation Regulates Human β-Cell Biology in Physiological States and in Type 2 Diabetes. (Jesus et al., 2019)
1. Could MeRIP-seq be subject to species restrictions?
For species with reference genomes, those genomes assembled at the chromosomal level, and those with comprehensive gtf annotation files, this methodology is applicable. There are distinct methods and parameters involved in peak-detection software for eukaryotes, prokaryotes, or viruses. Therefore, it is advisable for projects focusing on prokaryotes or viruses to conduct preliminary evaluations.
2. Why is it necessary to simultaneously measure Input and IP samples in MeRIP experiments?
The MeRIP project is characterized by two components: the Input and the IP. In the experimental procedure, the IP sample utilizes an m6A antibody to specifically enrich the methylated RNA fragments. In contrast, the Input, solely consisting of fragmented RNA, acts as a control to mitigate background noise. Both the IP and Input components are processed in parallel for library construction and sequencing. For peak detection analysis, data from both samples must be integrated, utilizing the Input data to exclude peaks corresponding to high basal expression levels or non-specific binding events, thereby maximizing the precision of calling peaks.
3. The strengths and limitations of MeRIP sequencing are what?
The MeRIP sequencing technique offers several advantages:
High-throughput: It facilitates the concurrent analysis of a substantial number of RNA samples. High Sensitivity: It is capable of detecting m6A modifications with low abundance. Spatial Resolution: This technique can accurately determine the location of m6A modifications.
However, the MeRIP sequencing technique also has limitations:
Antibody Dependent: The selection and quality of antibodies can potentially affect the experimental results.
Precision: While the sequencing data can provide a general location of m6A modifications, it isn't capable of pinpointing the precise base.
Complex Data Analysis: A high degree of specialized data analysis skills is required to interpret sequencing data and identify m6A modification sites.
4. What is the principle of MeRIP sequencing?
The principle of Merip sequencing is based on MeRIP technology and high-throughput RNA sequencing. Initially, RNA samples undergo MeRIP experiments, wherein specific antibodies are employed to bind RNA molecules modified with m6A. Subsequently, m6A-modified RNA molecules are enriched from total RNA via immunoprecipitation. Following enrichment, the RNA is subjected to reverse transcription and library preparation for DNA sequencing using high-throughput sequencing technology. Ultimately, sequencing data analysis is performed to determine the positions and relative abundances of m6A modifications.
RNA methylomes reveal the m6A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening
Journal: Genome Biology
Impact factor: 14.028
Published: 06 August 2019
Backgrounds
It is well-established that the modifications of DNA 5mC and mRNA m6A play critical roles in the regulation of gene expression. The conservation of the DNA 5mC modification underscores its fundamental role in modulating several biological processes. This study highlights its connection to the maturation of fruit. Prior investigations have shown that mutagenesis of the DNA demethylase gene SlDML2 in tomatoes triggers hypermethylation across the genome and significantly inhibits fruit maturation. However, the role that m6A plays in this process, as well as the potential interactions between 5mC and m6A, remain uncategorized. It is already known in Arabidopsis that m6A modification, mediated by the RNA demethylase SlALKBH2, can regulate the homologous gene of the DNA demethylase SlDML2.
Methods
- llumina HiSeq X, PE150
- Peak calling
- Differential methylation analysis
- Enrichment analysis of genes associated with peaks
Results
1. m6A modification displays a dynamic shift during the fruit ripening process, with overall m6A level gradually decreasing as the fruit ripens, mirroring DNA methylation patterns. In maturation defect mutations such as Cnr, alongside DNA hypermethylation, the overall m6A level is also higher.
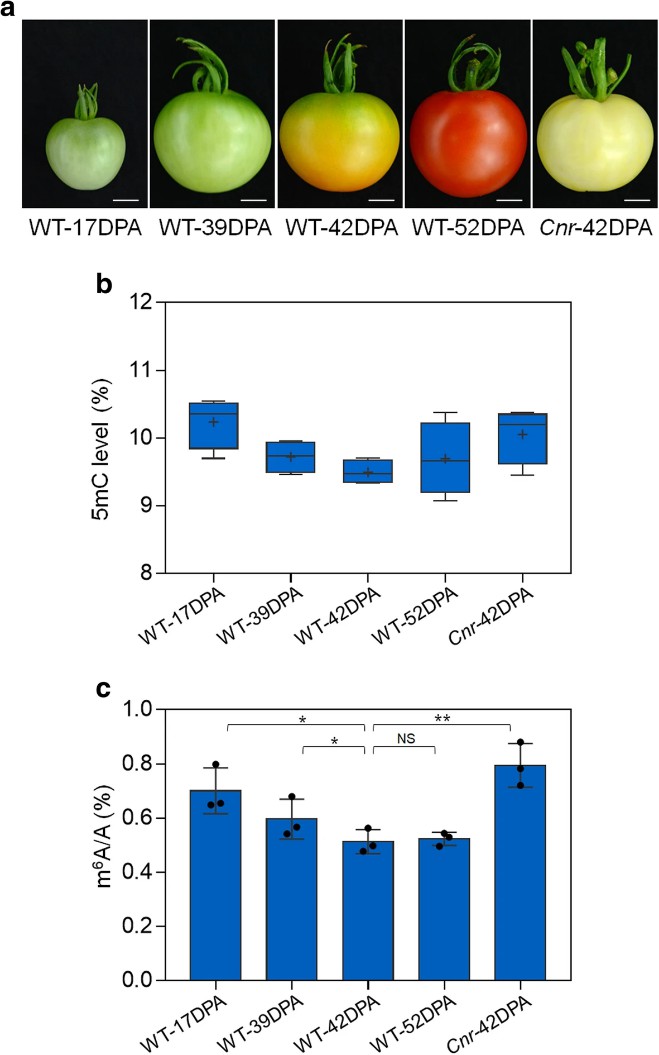 Figure1 Dynamics of DNA methylation (5mC) and mRNA m6A methylation in tomato fruit ripening.
Figure1 Dynamics of DNA methylation (5mC) and mRNA m6A methylation in tomato fruit ripening.
2. The methylation modification, m6A, is widely distributed in the mRNA of tomato fruits, exhibiting an inverse relationship with gene transcription levels.
The overall m6A levels during fruit ripening and in the Cnr mutant correspond to the expression of the m6A demethylase gene SlALKBH2, which is subject to DNA methylation regulation. SlALKBH2, an m6A demethylase, has the ability to bind to the mRNA of the DNA demethylase gene, SlDML2, in order to regulate its m'6A modification and stability. Upon mutation of the SlALKBH2 gene, increased levels of m6A in SlDML2 coincide with reduced mRNA content, thus impeding normal fruit maturation.
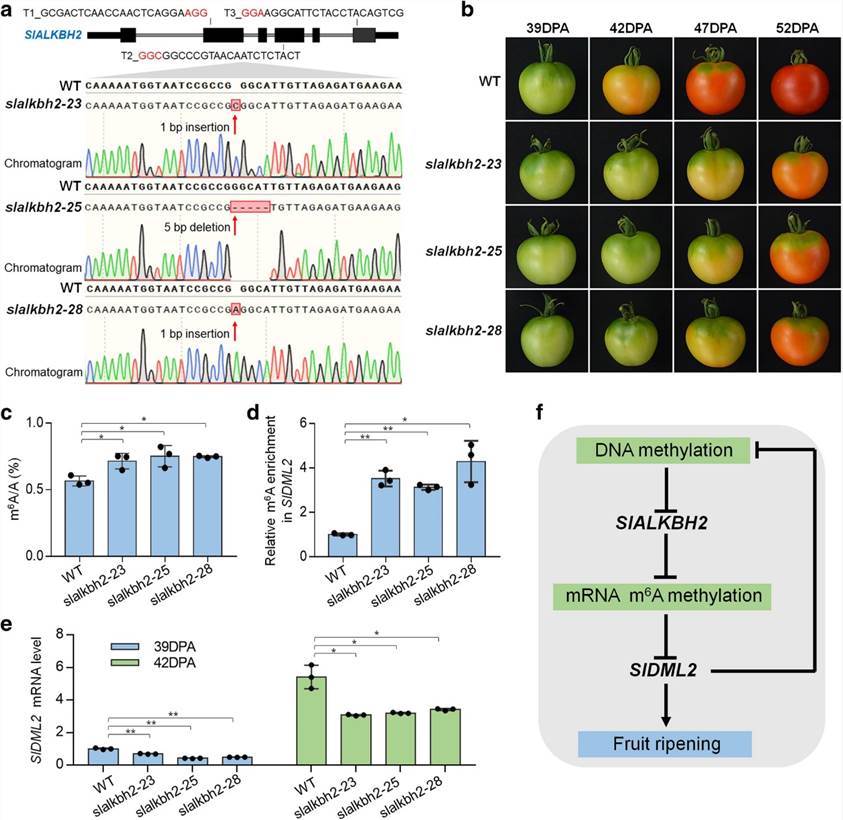 Figure2. SlALKBH2 is necessary for normal tomato fruit ripening.
Figure2. SlALKBH2 is necessary for normal tomato fruit ripening.
Conclusion
This study preliminarily confirms the connection between DNA methylation and RNA methylation, revealing a new mechanism of fruit ripening regulation, providing a new insight for elucidating the control network of fruit maturity. Given the multifunctionality of DNA methylation and RNA methylation, the feedback regulation mechanism demonstrated in this study will also apply to other biological processes.
Reference:
- Zhou L, Tian S, Qin G. RNA methylomes reveal the m 6 A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome biology, 2019, 20: 1-23.
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


