
 Sample Submission Guidelines
Sample Submission Guidelines
What is mRNA Sequencing
mRNA is transcribed from protein-coding genes and serves as the intermediate RNA molecule that transfers genetic information from DNA to proteins. In eukaryotes, DNA is initially transcribed into pre-mRNA, which undergoes a series of processing steps to form mature mRNA. The mature mRNA is then transported to the cytoplasm and translated by ribosomes into proteins. The maturation of mRNA involves three main processing events: splicing, 5' capping, and 3' polyadenylation. As a result, mature mRNA molecules possess a poly(A) tail at their 3' end.
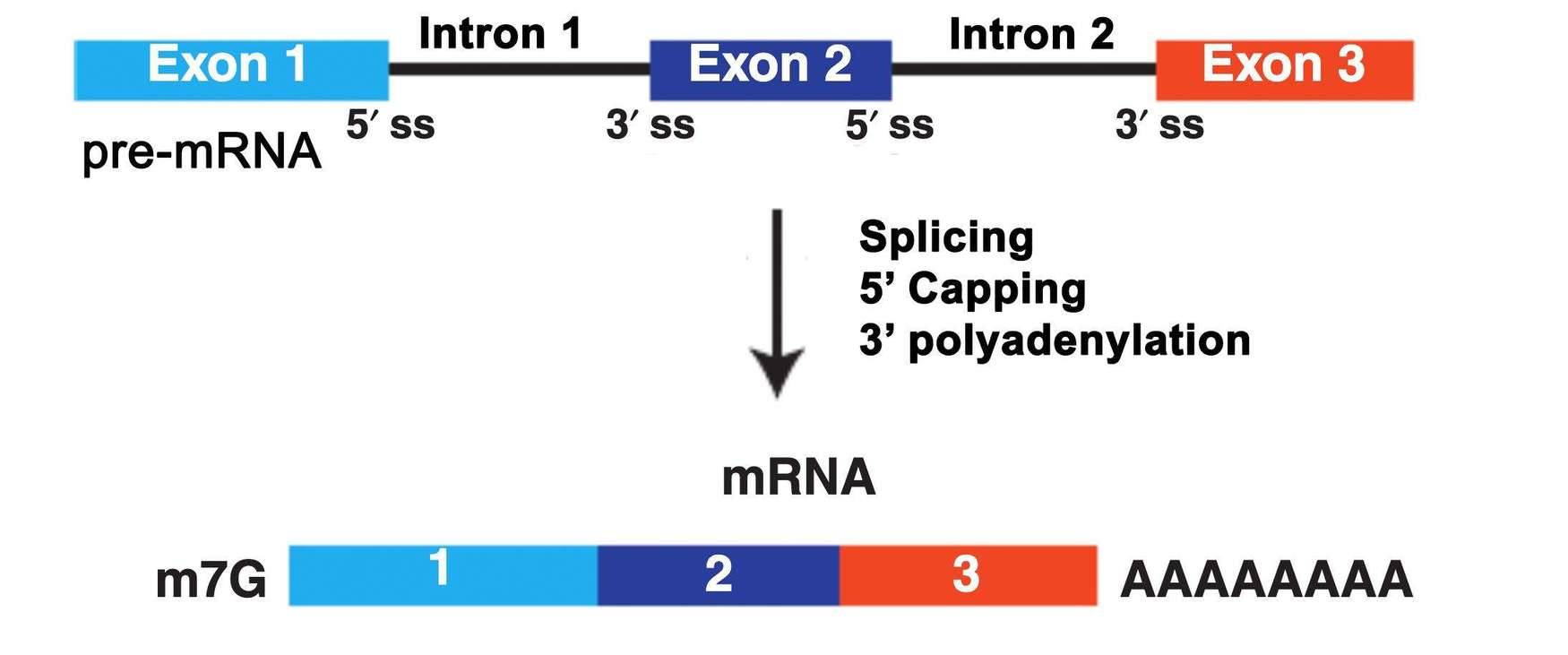 Fig 1. What is mRNA.
Fig 1. What is mRNA.
mRNA-seq is a high-throughput sequencing technology developed for detecting mRNA expression and processing. The study of mRNA sequencing in eukaryotes focuses on the collection of mRNAs transcribed from specific cells or tissues under certain functional states. It can be used to investigate differential mRNA expression, detect structural variations, and screen molecular markers associated with diseases or traits. It can also reveal the complexity of the transcriptome, determine gene and transcript structures, variable splicing, RNA editing, polyadenylated non-coding RNAs, and novel transcripts. Currently, mRNA-seq has been widely applied in fields such as basic research, molecular breeding, clinical diagnosis, and drug development.
mRNA-seq Technical Principle
For RNA containing poly(A) tails, such as mRNA and some lncRNAs, enrichment is performed using oligo dT, which captures the polyadenylated RNA, also known as PolyA-Seq. The enriched mRNA/lncRNA is then fragmented, reverse transcribed, ligated with common sequencing adapters, and PCR amplified to generate sequencing libraries. The resulting sequencing data is subjected to bioinformatics analysis to obtain information on gene expression, alternative splicing, RNA editing, and other features present in the RNA.
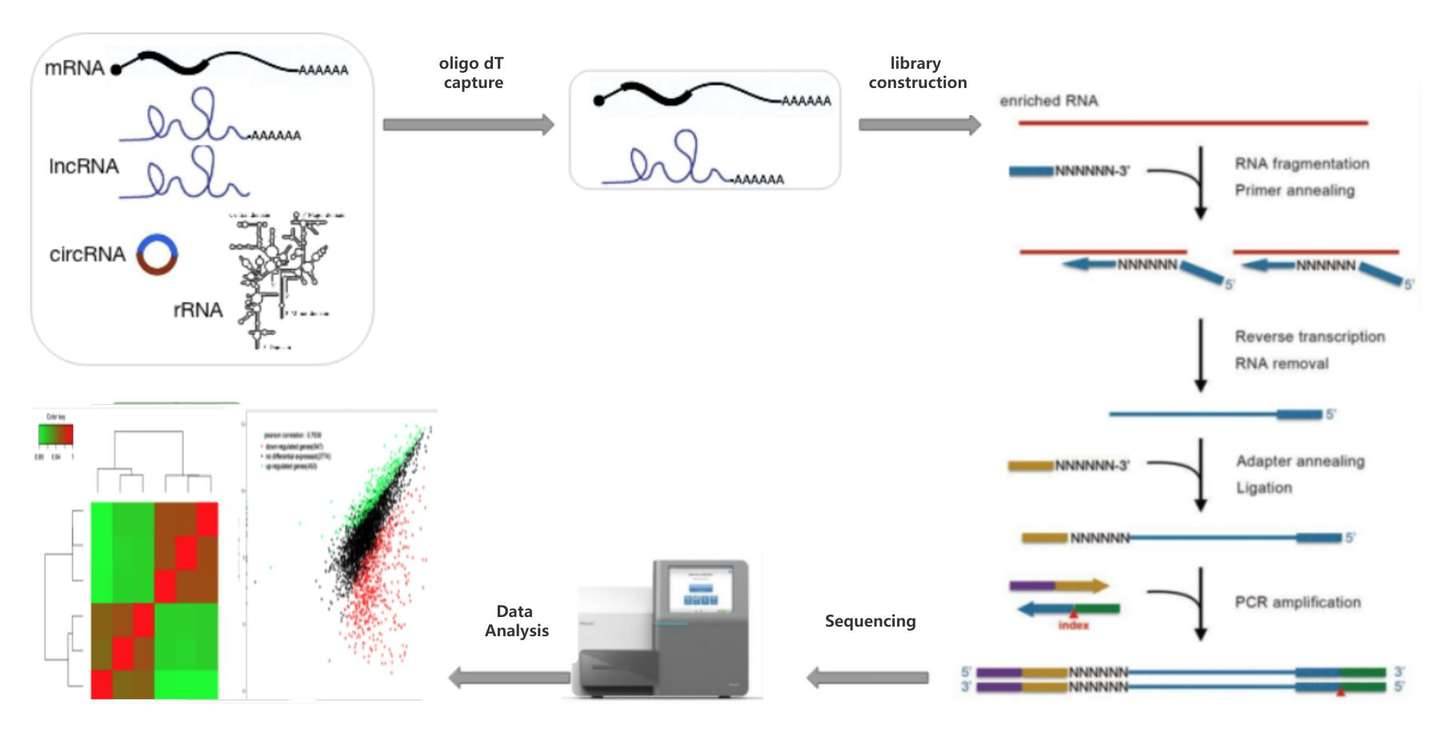 Fig 2. General workflow of mRNA-seq.
Fig 2. General workflow of mRNA-seq.
Advantages of our mRNA Sequencing Service
- Compared to gene expression microarrays, mRNA-Seq has several advantages in transcriptome analysis:
- It has a broader dynamic range, increasing both sensitivity and accuracy in measuring fold changes in gene expression.
- It can capture known features as well as novel features.
- It can be widely applied to various species.
- In addition to calculating gene expression levels, it can detect sequence and structural variations in transcripts.
- Increasing sequencing depth allows for a wider dynamic range of detection, enabling the identification and quantification of both highly abundant and low abundant transcripts within a span of six orders of magnitude.
- In addition to detecting the expression levels of known transcripts, it can also discover novel transcripts.
- When applied to species without a reference genome, it can discover new genes through de novo assembly.
mRNA Sequencing Service Workflow
For mRNA-Seq, our company provides experimental services including but not limited to library construction and sequencing based on tissues and cell lines, as well as streamlined and customized bioinformatics analysis and interpretation services.

Service Specification
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Data Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
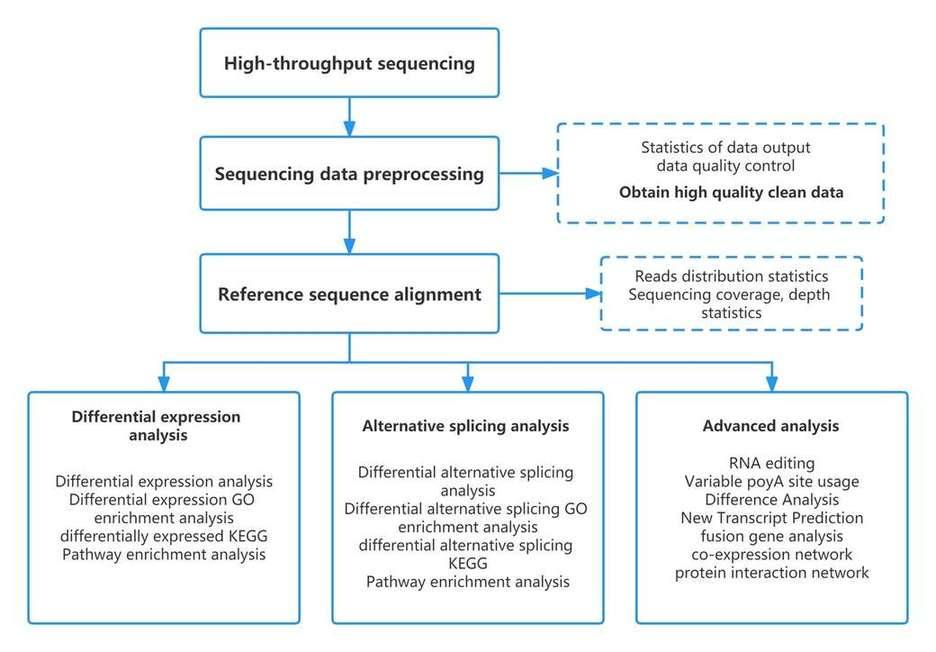
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in mRNA Sequencing for your writing (customization)
At CD Genomics, we offer cutting-edge mRNA sequencing services designed to cater to diverse research needs. Our service includes rigorous sample quality control, expert library preparation, high-throughput sequencing, and detailed data analysis. We prioritize delivering accurate and comprehensive results that align with the specific goals of your research. Get in touch with us to explore how our mRNA sequencing solutions can support and enhance your scientific endeavors.
Partial results are shown below:

Sequencing quality distribution

A/T/G/C Distribution

IGV Browser Interface

Correlation Analysis Between Samples

PCA Score Plot

Venn Diagram

Volcano Plot

Statistics Results of GO Annotation

KEGG Classification
1. Can all eukaryotes undergo mRNA sequencing?
Eukaryotic mRNA contains a poly-A tail, and the common method for enriching eukaryotic mRNA involves capturing mRNA from total RNA using magnetic beads attached to poly-T oligos, followed by library construction for sequencing. Therefore, theoretically, all eukaryotes can undergo mRNA sequencing.
2. Why does mRNA sequencing require higher RNA integrity than whole transcriptome sequencing?
In mRNA sequencing library construction, RNA molecules with a poly-A tail are captured using magnetic beads. If RNA integrity is poor and mRNA is partially degraded, leading to poly-A tail loss, the captured mRNA from samples with lower integrity is reduced, resulting in lower gene coverage. Hence, a higher level of RNA integrity is required.
3. Can mRNA-seq detect non-coding RNA?
mRNA-seq primarily captures RNA molecules with a poly(A) tail; hence, theoretically, any poly(A)-tailed molecule expressed in the cell can be detected. This includes mRNA and long non-coding RNAs with poly(A) tails.
4. What is the difference between total RNA-seq and mRNA-Seq?
Total RNA-Seq, or whole transcriptome sequencing, is an exhaustive method that encompasses the sequencing of all RNA species after ribosomal RNA (rRNA) removal, including coding and non-coding RNAs. This encompasses RNA like rRNA, precursor mRNA (pre-mRNA), messenger RNA (mRNA), and a variety of non-coding RNAs (ncRNA) such as transfer RNA (tRNA), microRNA (miRNA), and long non-coding RNA (lncRNA). Conversely, mRNA-Seq specifically targets coding regions by enriching polyadenylated (poly(A)) RNA, making it a more cost-effective and streamlined approach for studies primarily focused on mRNA analysis.
5. Should you choose mRNA sequencing or total RNA sequencing?
For research involving eukaryotic organisms and with a primary interest in coding regions, mRNA-Seq is the optimal choice. The mRNA-Seq approach enriches mRNA through Poly(A) selection, reducing sequencing costs and complexity, particularly suitable for samples with limited starting material. On the other hand, if a comprehensive analysis encompassing all RNA, including coding and non-coding RNA molecules, is required, total RNA-Seq is the preferred option, despite its higher cost and greater sequencing data demand. The selection between methods should be based on specific experimental objectives, biological questions, sample types, and budget constraints.
Molecular mechanism underlying cadmium tolerance differentiation in Lentinula edodes as revealed by mRNA and milRNA analyses
Journal: Journal of Hazardous Materials
Impact factor: 14.224
Published: 15 October 2022
Background
Cadmium (Cd) is a widely distributed toxic heavy metal capable of inducing several diseases in the human body, such as breast cancer and kidney cancer. With the rapid development of industrial and agricultural activities, cadmium contamination has become a serious global issue. Therefore, it is essential to remediate Cd-contaminated areas. Fungal remediation is a novel and important restoration technique, yet the molecular mechanisms underlying fungal responses to heavy metal stress remain unclear. This study conducted transcriptome sequencing to analyze differential mRNA and miRNA expression between Cd-tolerant (YS119) and Cd-sensitive (YS45) mushrooms. The findings provide a theoretical basis for further exploration of the molecular mechanisms of cadmium tolerance in fungi and help elucidate the potential roles of miRNAs in fungal responses to heavy metal stress.
Materials & Methods
- Cd-tolerant mushroom YS119
- Cd-sensitive mushroom YS45
- RNA extraction
- mRNA sequencing
- miRNA-seq
- PCA and correlation analysis
- DEGs analysis
- miRNA target gene prediction
- GO and KEGG analysis
- Integrated analysis of mRNAs and miRNAs
Results
1. mRNA Sequencing Analysis
A total of 577,586,098 raw reads were obtained from all samples, resulting in 577,165,672 clean reads after filtration. Pearson correlation coefficients and Principal Component Analysis (PCA) indicated a high reproducibility between biological replicates of the samples, enabling further in-depth analysis (Figure 1A, B).
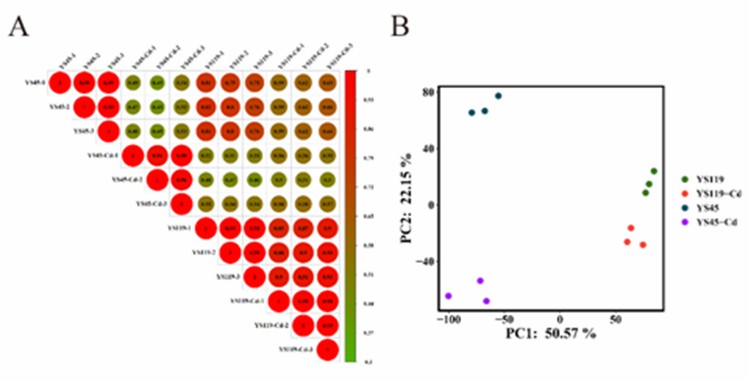 Figure 1 Pearson correlation coefficient and PCA analysis.
Figure 1 Pearson correlation coefficient and PCA analysis.
Under Cd stress, YS45 exhibited a stronger response compared to YS119, with 2036 differentially expressed genes (DEGs) in YS45 (1034 upregulated, 1002 downregulated) versus 370 DEGs in YS119 (239 upregulated, 131 downregulated). GO and KEGG analyses revealed that both mushroom varieties had 170 commonly upregulated DEGs, enriched in 14 GO categories and one KEGG pathway, and 107 commonly downregulated DEGs, enriched in 12 GO categories and one KEGG pathway, indicating that genes involved in protein stability, ion binding, and redox activity play roles in Cd stress response. Specifically, YS45 had 863 uniquely upregulated genes enriched in 20 GO categories and 12 KEGG pathways, while YS119 had 64 uniquely upregulated genes enriched in 11 GO categories and one KEGG pathway.
Additionally, YS45 had 890 uniquely downregulated genes enriched in 22 GO categories and 8 KEGG pathways, whereas YS119 had 23 uniquely downregulated genes enriched in 7 GO categories with no KEGG pathway enrichment. This suggests that YS45's severe suppression of genes related to cell wall remodeling, DNA repair, sugar metabolism, and proteolysis under Cd stress may reduce its Cd tolerance, with differential expression of genes involved in transcription regulation, transport, lipid metabolism, metal binding, glutathione metabolism, and redox homeostasis contributing to the tolerance differences between the two varieties.
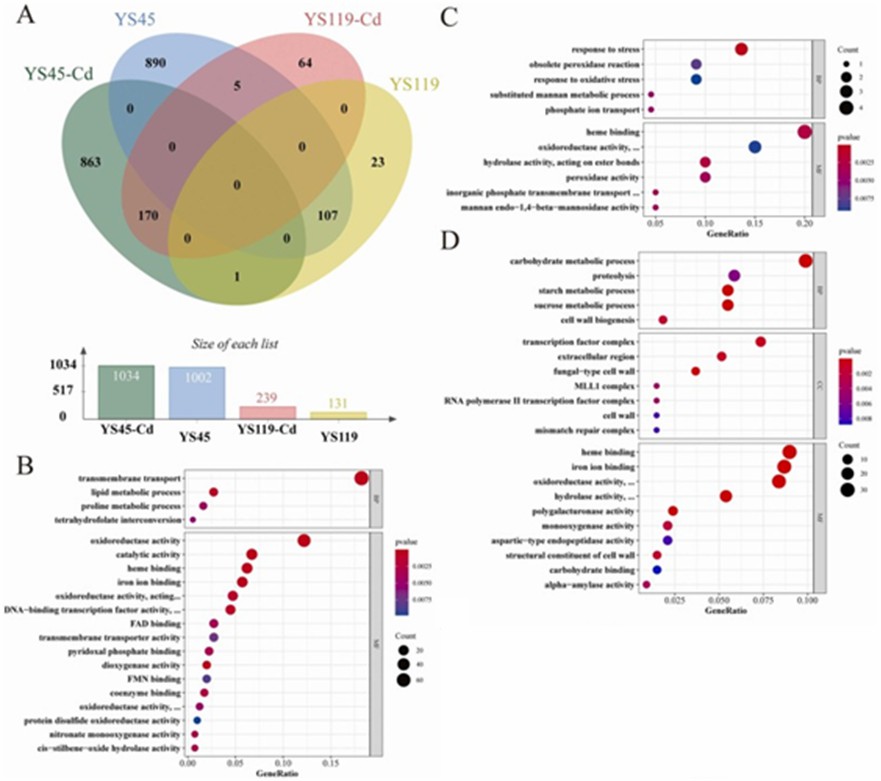 Figure 2 DEGs analysis.
Figure 2 DEGs analysis.
2. Integrated analysis of mRNA and miRNA
miRNAs regulate gene expression by degrading or inhibiting their target mRNAs. In YS45, among 1620 DE miRNAs' targets, 216 are DEGs, with 111 showing opposite expression trends to the miRNAs (Figure 3A, B). Genes potentially negatively regulated by these miRNAs include those encoding TFs, transport proteins, cytochrome P450s, DNA repair proteins like MutL, proteases, glutathione S-transferases, and carbohydrate-active enzymes (CAZymes). In YS119, out of 525 DE miRNAs' targets, only 14 are DEGs, with 3 showing opposite expression trends (Figure 3C, D), encoding glycoside hydrolase, cellulose growth protein, and retinol dehydrogenase.
These results suggest that miRNAs may not significantly affect the expression of most target genes in mushrooms. mRNA degradation may not be the primary regulatory mechanism; instead, translational inhibition and competition with endogenous RNAs for miRNA binding could play key roles.
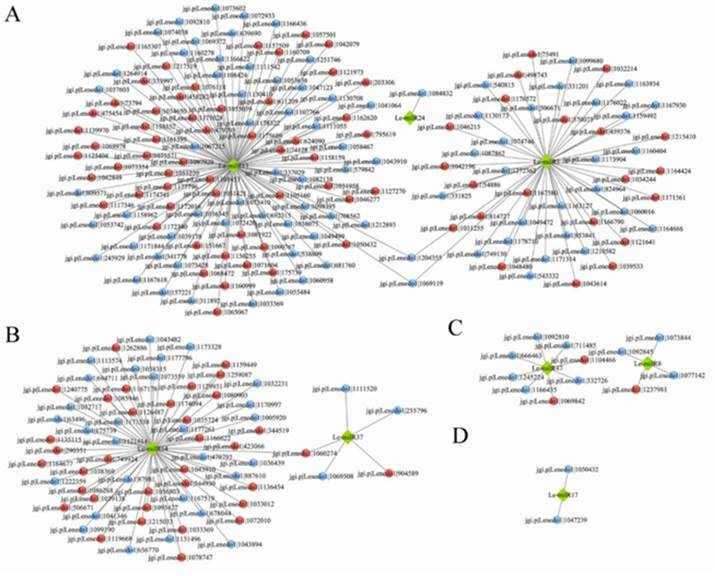 Figure 3 Integrated analysis of mRNA and miRNA.
Figure 3 Integrated analysis of mRNA and miRNA.
Conclusion
This study delves into the notable molecular mechanisms underlying the differences in cadmium (Cd) tolerance between two mushroom varieties, YS45 and YS119. Under Cd stress, YS45 exhibited greater protein aggregation compared to YS119, impacting normal cellular activities and diminishing the mushroom's Cd tolerance. Comparative analyses of mRNA and miRNA revealed the involvement of genes or miRNAs associated with cell wall remodeling, transport, metal chelation, redox homeostasis, signal transduction, transcriptional regulation, lipid and carbohydrate metabolism, protein hydrolysis, DNA repair, and cell cycle regulation in the modulation of Cd tolerance. Furthermore, the overexpression of the LeAmy gene confirmed its role in enhancing Cd tolerance in mushrooms. The findings lay a theoretical groundwork for a deeper understanding of the molecular mechanisms behind the differences in Cd tolerance in mushrooms, aiding in the development of heavy-metal-tolerant fungal cultivars.
Reference:
- Shen N, Xu C, Zhang J, et al. Molecular mechanism underlying cadmium tolerance differentiation in Lentinula edodes as revealed by mRNA and milRNA analyses. Journal of Hazardous Materials, 2022, 440: 129841.
Here are some publications that have been successfully published using our services or other related services:
Human Immature Dental Pulp Stem Cells Did Not Graft into a Preexisting Human Lung Adenocarcinoma
Journal: Case Reports in Oncology
Year: 2022
Circular DNA tumor viruses make circular RNAs
Journal: Proceedings of the National Academy of Sciences
Year: 2018
Repeated immunization with ATRA-containing liposomal adjuvant transdifferentiates Th17 cells to a Tr1-like phenotype
Journal: Journal of Autoimmunity
Year: 2024
Role of the histone variant H2A.Z.1 in memory, transcription, and alternative splicing is mediated by lysine modification
Journal: Neuropsychopharmacology
Year: 2024
FAK loss reduces BRAFV600E-induced ERK phosphorylation to promote intestinal stemness and cecal tumor formation
Journal: Elife
Year: 2023
Identification of circular RNAs regulating cardiomyocyte proliferation in neonatal pig hearts
Journal: JCI insight
Year: 2024
See more articles published by our clients.


