
 Sample Submission Guidelines
Sample Submission Guidelines
What are WES, WGS and Panel Sequencing
Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES) are abbreviated terms for sequencing the entire genome and the entire exome, respectively. In the context of Next-Generation Sequencing (NGS), the term "Panel" represents a "gene package" or a set of targeted genes.
Characteristics of WGS, WES, and Panel Sequencing
WGS, WES, and Panel each exhibit distinct characteristics in the realm of genomics.
To illustrate, the entire genome sequence comprises approximately 3 gigabases (Gb). If sequencing data amounts to 10 Gb, it can cover the entire genome approximately 3 times, referred to as 3X sequencing depth. In contrast, the coding exons, constituting only 1% of the genome or around 30 megabases (Mb) in size, can achieve sequencing depths of over 100X with 10 Gb of sequencing data. This is even more pronounced for Panels targeting specific genes, where 1 Gb of data can achieve depths of around 1000X. Reduced data volume correlates with lower costs, but one significant advantage of Panels lies in their high sequencing depth.
What Are the Benefits of High Sequencing Depth?
Consider a wild-type locus with a GG genotype. If a heterozygous mutation occurs, the genotype becomes GT. Sequencing data will simultaneously display sequences containing both G and T, roughly in a 1:1 ratio. In genetic testing, sequencing data is used to infer the true genotype. To confidently conclude a heterozygous mutation at a given locus and prevent interference from sequencing errors, a certain quantity of variant sequences is required, necessitating a minimum sequencing depth, such as 20X.
Due to systematic biases in the sequencing and analysis processes, the distribution of sequences with G and T may not be a perfect 1:1 ratio. Additionally, the sequencing depth at different loci may vary, influenced by factors such as genomic GC content and probe specificity during capture. Higher sequencing depth mitigates these issues, reducing the likelihood of missing variant information in problematic regions.
Moreover, a substantial sequencing depth proves beneficial in identifying mosaic variations. In instances where variations manifest during the fertilization stage, irrespective of the number of cells subjected to sequencing, the variant sequences will each constitute approximately half. On the contrary, variations arising in intermediate developmental stages may exhibit mosaic patterns. The ability to discern lower proportions of mosaicism necessitates increased sequencing depth, making gene Panels particularly adept at this task.
Summary of Characteristics Across Three Sequencing Technologies
Gene Panels showcase the ability to achieve elevated sequencing depth, conferring an advantageous position in genetic analysis. Moreover, they offer substantial customization flexibility, facilitating the inclusion of pathogenic regions within non-coding domains.
WES, strategically positioned between WGS and Panel sequencing, strikes a harmonious balance by delivering an expanded sequencing scope at a generally acceptable cost. This technology empowers researchers to attain comprehensive sequencing coverage while sustaining a commendable sequencing depth.
WGS stands out for its unparalleled sequencing scope, spanning the entire genome. However, to detect base pair variations, a substantial dataset of approximately 100 gigabases (Gb) is requisite. The current challenges associated with the feasibility and affordability of acquiring such extensive data underscore the considerations for its widespread adoption.
How to Choose Panel, WES or WGS
The ongoing debate in the realm of genetic testing revolves around the selection of Panel, WES, or WGS. Each method boasts distinctive advantages, prompting the need for a thoughtful decision-making process.
In cases where testing objectives are clearly defined, the initial preference should be towards Panel sequencing to ensure heightened sensitivity within the designated detection range. Conversely, when testing goals are less defined, considerations should shift towards WES or WGS to uncover a broader spectrum of pathogenic factors. WGS involves sequencing every base pair of the genome, while WES and Panel sequencing employ targeted sequencing techniques.
Ultimately, the decision-making process is contingent upon the clarity of testing objectives. Opting for Panel sequencing guarantees precision in detecting known targets, whereas WES or WGS may be favored when the goal is to explore a wider array of potential pathogenic factors, particularly in scenarios where testing targets lack specificity.
Our Panel Sequencing Services
CD Genomics offers ccurate and cost-effective predesigned and custom NGS panel services, which involves capturing DNA fragments from multiple relevant gene target regions using specific gene capture probes. Subsequently, the captured DNA sequences in the target regions are determined using NGS technology, enabling the identification of target genes and mutation sites.
RNA Sequencing Panel
Targeted RNA sequencing is the optimal choice for studying gene expression and rearrangements, even with samples of poor quality such as FFPE and cfRNA. The probe technology targeting all exonic regions of genes of interest has high coverage, enabling comprehensive gene expression analysis, including subtype analysis of total RNA sequencing.
Exome Sequencing Panel
Exome sequencing panels, as a widely used method in disease research, can rapidly and effectively detect pathogenic gene variations in target genes across the genome. This approach enhances data coverage depth, effectively reducing data analysis time and sequencing costs.
Targeted Methylation Sequencing Panel
CD Genomics has introduced a probe technology based on bisulfite conversion, enabling methylation analysis of various sample types, such as gDNA and cfDNA. This method accurately analyzes the methylation status of target genes, providing comprehensive methylation results for research. Subsequently, CD Genomics also offers comprehensive bioinformatics analysis services for methylation data.
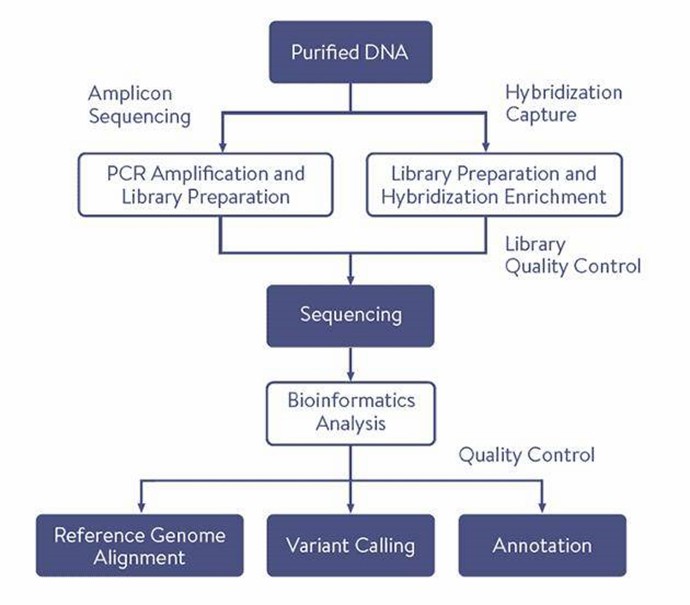
Why Choose Our Custom NGS Sequencing Panels
- Accuracy: Based on deep sequencing technology, the accuracy of results exceeds 99%.
- Specific Capture: Demonstrates excellent specificity, allowing for chromosome-specific capture in polyploid organisms.
- Flexible Customization: Highly adaptable, capable of simultaneously conducting gene sequence detection and targeted mutation detection in a single pipeline.
- High Detection Efficiency: Enables parallel sequencing of hundreds of thousands to millions of DNA molecules in a single run, facilitating the simultaneous detection of multiple diseases, numerous genes, and tens of thousands of mutation sites. This ensures rapid, efficient, reliable results with high throughput and cost-effectiveness.
Application of Gene Panel Sequencing
- Genetic Research: Discover novel variants, study gene functions, and understand biological processes and disease mechanisms.
- Disease Association Studies: Identify genes linked to genetic disorders or complex diseases and explore genetic variations in different populations.
- Cancer Research: Profile cancer-related mutations, classify cancer subtypes, and investigate tumorigenesis.
- Pharmacogenomics: Analyze genetic factors influencing drug responses and identify markers for drug toxicity.
- Functional Validation: Validate gene functions and study interactions within cellular pathways.
- Biomarker Discovery: Identify diagnostic and prognostic genetic markers for disease detection and progression.
- Rare Disease Investigation: Find genes associated with rare disorders and study genetic inheritance patterns.
Gene Panel Sequencing Workflow
CD Genomics' Gene Panel Sequencing Service offers a precise and efficient solution for targeted genetic research. The service encompasses custom gene panel design, high-throughput sequencing, and detailed data analysis to uncover important genetic variants relevant to your research objectives.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
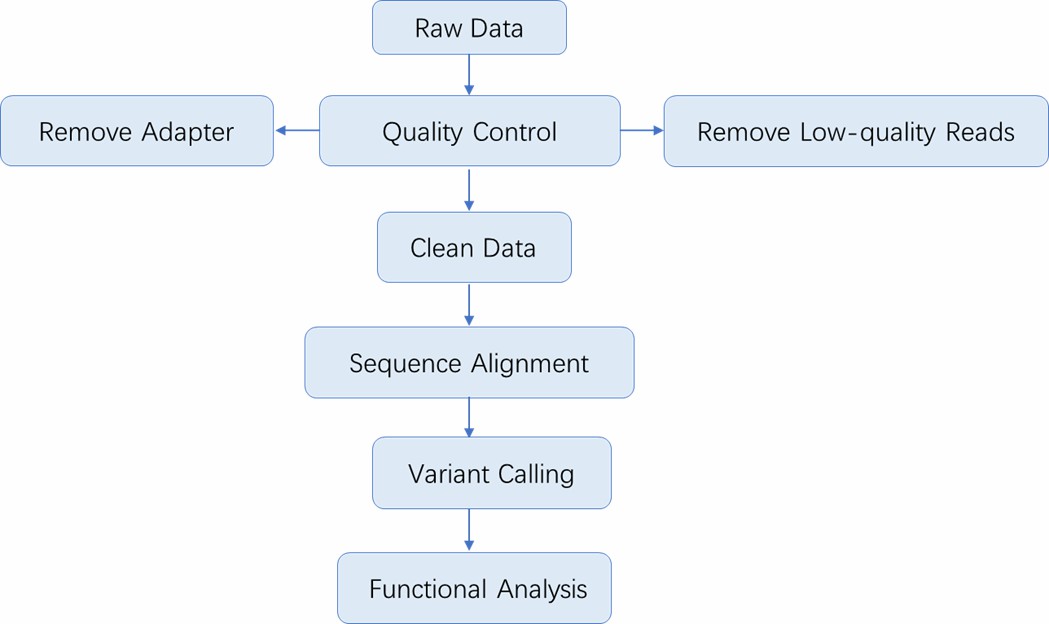
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Gene Panel Sequencing for your writing (customization)
Partial results are shown below:
1. When selecting genetic testing options, what do "small panel" and "large panel" mean?
In the context of NGS, "small panel" and "large panel" refer to different scopes based on the number of genes being analyzed.
Small Panel: This typically includes a set of genes ranging from a dozen to a few dozen. These panels generally focus on key driver genes associated with approved or clinically relevant targeted therapies for specific cancer types. They may also include a selection of tumor suppressor genes that have garnered significant research interest.
Large Panel: These panels encompass hundreds to thousands of genes. They not only cover driver genes pertinent to targeted therapies but also incorporate a broad spectrum of genes associated with cancer, as allowed by current research and technological capabilities. The results from large panels extend beyond the identification of targeted therapies for specific cancers to include cross-cancer drug options, immunotherapy-related markers such as Tumor Mutational Burden (TMB) and Microsatellite Instability (MSI), and potentially hereditary cancer genes.
2. How does Gene Panel Sequencing differ from Whole-Genome Sequencing?
Gene Panel Sequencing focuses on predetermined sets of genes or genomic regions, delivering high-resolution data for those targeted areas. In contrast, Whole-Genome Sequencing entails the examination of the entire genome, encompassing both coding and non-coding regions. This approach offers a comprehensive landscape of all genetic variations within an organism.
3. How do I choose the right gene panel for my research?
Consider the specific genes or pathways of interest, the research objectives, and any prior knowledge of the genetic factors involved. Consult with a genomic specialist or review available panels to ensure alignment with your research goals.
Identification of mutation gene prognostic biomarker in multiple myeloma through gene panel exome sequencing and transcriptome analysis in Chinese population
Journal: Computers in Biology and Medicine
Impact factor: 7.7
Published: September 2023
Background
Multiple myeloma (MM) is a prevalent blood cancer that primarily affects older adults and is less common in children in China. It shows regional variations in incidence and has a five-year survival rate below 40%. Effective treatments are currently lacking. NGS is proving valuable for understanding MM by identifying critical mutations. This study employs exome sequencing in 50 Chinese MM patients to uncover significant genetic changes and potential new targets for treatment.
Materials & Methods
Sample Preparation
- Clinical samples
- Bone marrow specimens
Sequencing
- Targeted gene panel
- Whole-exome sequencing
- Illumina MiSeq
- DEG identification
- GO and KEGG enrichment analysis
- Survival analysis
- Statistical analysis
Results
Exome sequencing of 50 MM patients targeting 400 genes revealed various mutation types, with nonsynonymous single nucleotide variants (SNVs) being predominant. The most common mutations were C > T substitutions, and each sample had a median of 576 mutations. The top ten frequently mutated genes were MUC16, MUC4, TTN, AHNAK2, MUC17, OBSCN, OR4C3, MUC2, MKI67, and PRUNE2, all showing 100% mutation frequency.
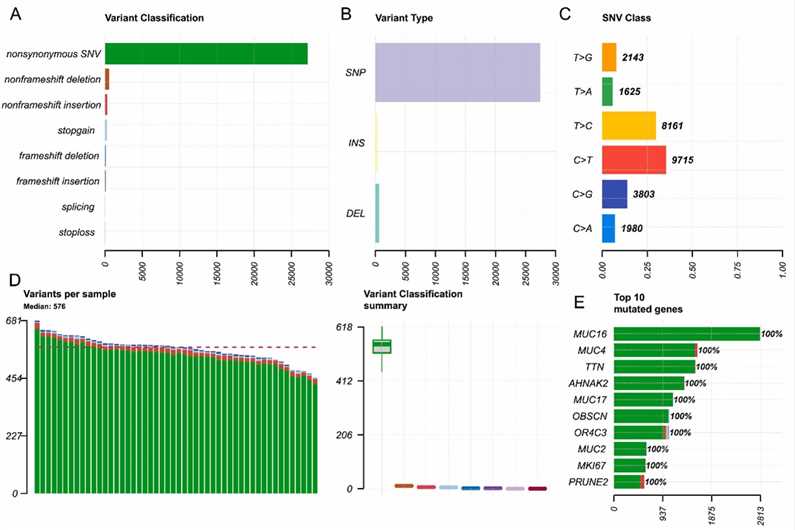 Fig. 1. Overview of the mutation status of the 50 patients with MM.
Fig. 1. Overview of the mutation status of the 50 patients with MM.
The study analyzed the mutational landscape of 50 MM patients using a 400-gene panel. It identified mutations in 337 genes, with 48 genes showing 100% mutation frequency and 31 more than 90%. Key mutated genes include CACNA1I, ID3, and EGFR. Functional enrichment revealed that mutant genes were associated with DNA modification, extracellular matrix functions, and several key signaling pathways, including PI3K-Akt and Notch.
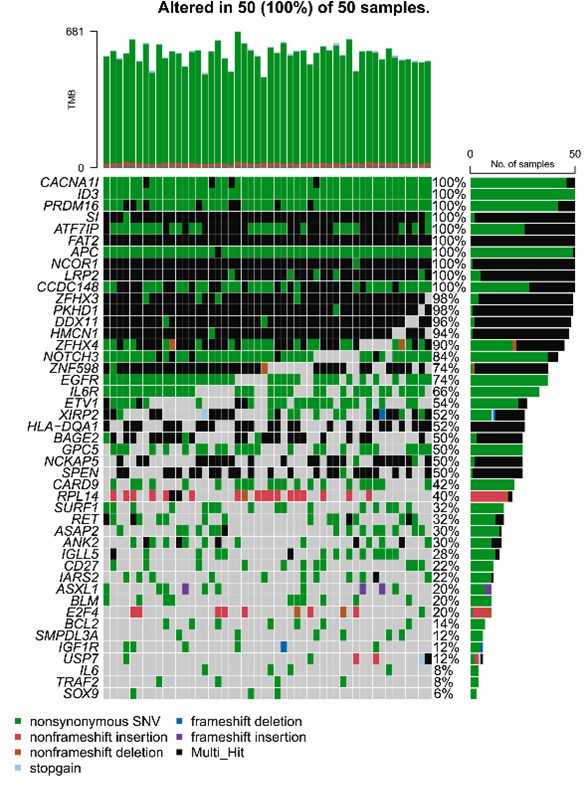 Fig. 2. The common 45 mutated genes of 50 patients were visualized by OncoPrinter.
Fig. 2. The common 45 mutated genes of 50 patients were visualized by OncoPrinter.
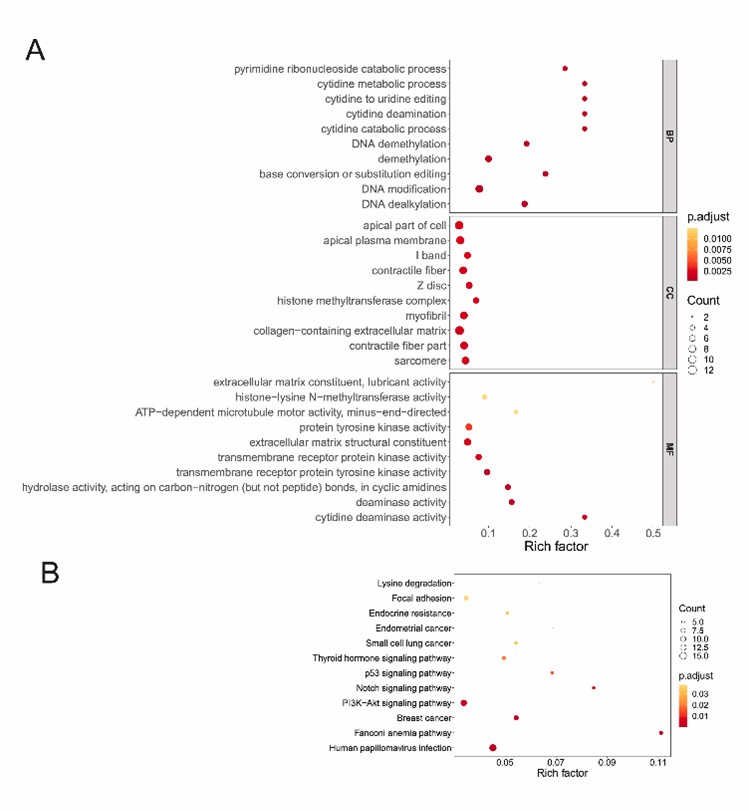 Fig. 3. Function enrichment analysis of mutant genes in MM.
Fig. 3. Function enrichment analysis of mutant genes in MM.
Gene expression analysis from the GSE6477 dataset identified 1247 differentially expressed genes (DEGs), with 660 downregulated and 587 upregulated. Notable DEGs include RNASE2 and RPS19. Analysis of common genes between mutant genes and DEGs highlighted 33 genes, including ID3 and MYC, which were enriched in pathways related to cell proliferation, myeloid differentiation, and viral carcinogenesis.
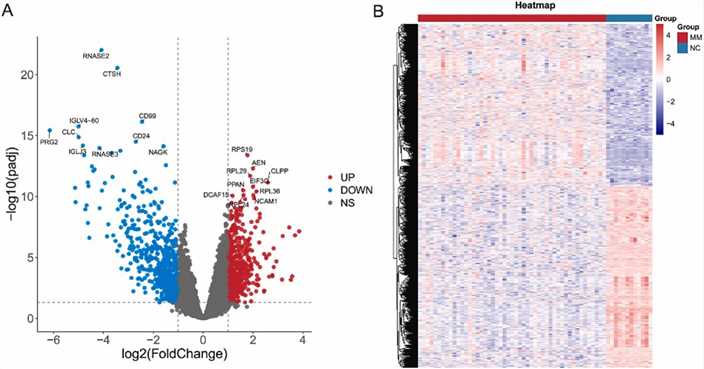 Fig. 4. Identification of DEGs in GSE6477 datasets.
Fig. 4. Identification of DEGs in GSE6477 datasets.
Conclusion
Multiple myeloma (MM) in China shows a low survival rate. Sequencing of 50 patients found mutations in 337 genes, notably MUC16, MUC4, and TTN. Key mutations in BCL6 and BIRC3 are linked to worse outcomes and immune changes, offering new treatment targets but needing further research.
Reference
- Xie C, Zhong L, Luo J, et al. Identification of mutation gene prognostic biomarker in multiple myeloma through gene panel exome sequencing and transcriptome analysis in Chinese population. Computers in Biology and Medicine, 2023, 163: 107224.
Here are some publications that have been successfully published using our services or other related services:
Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Journal: Cell Death & Disease
Year: 2024
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
See more articles published by our clients.


