
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics offers cutting-edge Nanopore RNA Methylation Sequencing services to reveal m6A modification patterns directly from RNA molecules. Our high-throughput method allows for single-base resolution analysis, providing detailed insights into RNA epigenetics. This service supports investigations into gene regulation, RNA metabolism, and disease mechanisms.
What is Nanopore RNA Sequencing
DNA Methylation and RNA methylation are processes facilitated by methyltransferases, where a methyl group (CH3) is added to a specific atom on a DNA or RNA molecule. In cellular RNA, over 100 types of chemical modifications have been identified. Various RNA methylation modifications include m6A RNA methylation, m5C RNA methylation, m1A RNA methylation, m7G RNA methylation, and more. Among these, the most prominent and extensively studied is m6A RNA methylation, occurring at the sixth nitrogen atom of the adenine residue in RNA molecules (N6-methyladenosine, m6A). This modification represents the most common post-transcriptional modification in eukaryotic mRNA, constituting approximately 80% of all RNA methylation modifications.
ONT RNA methylation sequencing is carried out using nanopore technology. Nanopores are small pores with a diameter in the nanometer range. When a single strand of RNA passes through a nanopore, it generates changes in electrical current. These current fluctuations can be precisely recorded and deciphered by specific algorithms to reveal the sequence information of RNA along with its methylation modifications. The key aspect of ONT technology lies in its ability to directly sequence long RNA molecules and detect various modifications on RNA, including N6-methyladenine (m6A), 5-methylcytosine (m5C), and others.
Methods for Detecting RNA Methylation
MeRip-seq (Methylated RNA Immunoprecipitation):
MeRip-seq is based on the principle of antibody-specific binding to methylated bases. It utilizes m6A-specific antibodies for immunoprecipitation to enrich RNA fragments that undergo methylation. High-throughput sequencing is then employed to detect transcripts with m6A modifications. However, this method can only identify regions with high methylation and lacks single-base resolution in recognizing RNA methylation.
Nanopore Sequencing Technology:
Nanopore sequencing is a long read sequencing technology based on electric signal recognition of base sequences. Different modifications on RNA bases cause variations in the obstruction size as they pass through a nanopore channel, generating characteristic electrical signals. Real-time monitoring of these signals allows for the determination of the corresponding base types and whether they carry base modifications. In other words, Nanopore sequencing can directly perform full-length transcriptome sequencing on natural RNA samples without the need for specific antibody binding. It enables the detection of m6A (N6-methyladenosine) methylation modifications on RNA with single-base resolution, while simultaneously providing quantitative gene/transcript expression and transcript structure identification.
Principles of Nanopore RNA Methylation Sequencing
Nanopore sequencing technology leverages the detection of signals generated when a single molecule passes through a nanopore, causing a potential difference on either side of the pore. The nanopore's diameter allows only individual nucleotide polymers to pass through. The charged nature of different bases, including those with methylated modifications, varies, enabling the detection of corresponding base types and methylation information through differences in electrical signals.
Advantages of Nanopore RNA Methylation Sequencing Service
- Single Nucleotide Resolution Across the Entire Transcriptome: Identification of m6A methylation modifications at single nucleotide resolution throughout the transcriptome.
- Direct Reading of m6A Methylation Information: Direct extraction of m6A methylation information without the need for immunoprecipitation enrichment experiments.
- Simultaneous Quantification of Gene/Transcript Expression and Transcript Structure: Conduction of both quantitative analysis of gene/transcript expression and investigation of transcript structure concurrently.
- Absence of Reverse Transcription and PCR Bias: Nanopore direct RNA sequencing eliminates the need for disruption, reverse transcription, and PCR amplification, allowing for the direct full-length transcriptome sequencing of natural RNA samples.
Applications of Nanopore RNA Methylation Sequencing
- Investigating the functional role of molecular participants involved in RNA methylation processes during disease or specific life processes.
- Unraveling the alterations in RNA methylation modifications during disease or particular life processes, thereby elucidating disease-specific m6A RNA methylation patterns.
- Exploring the relationship between specific RNA methylation modifications and disease during disease or specific life processes.
- Identifying novel molecules involved in RNA methylation modifications or establishing new methodologies for the study of RNA methylation modifications.
Nanopore RNA Methylation Sequencing Workflow
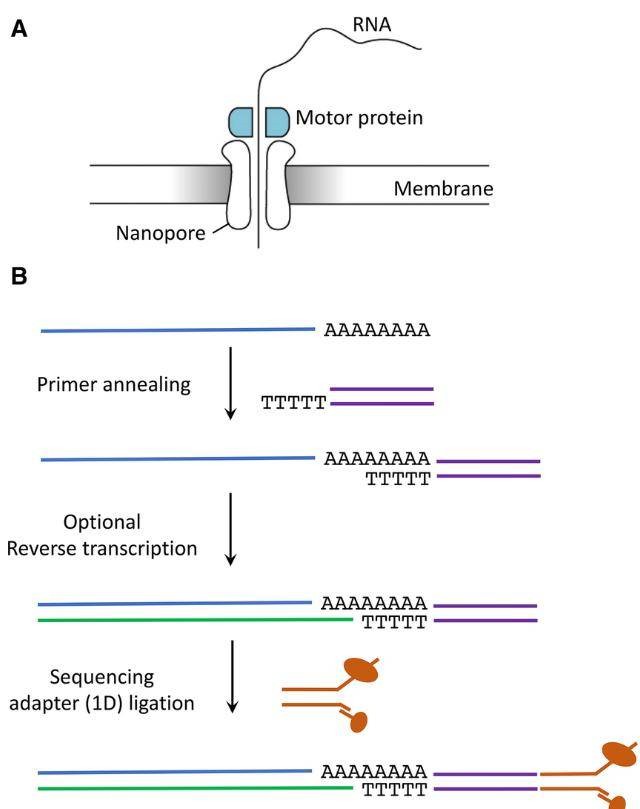 Fig 1. Library preparation steps using Oxford Nanopore Technologies. (Nicky Jonkhout et al,. 2017)
Fig 1. Library preparation steps using Oxford Nanopore Technologies. (Nicky Jonkhout et al,. 2017)
Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses: m6A RNA Methylation Detection:
|
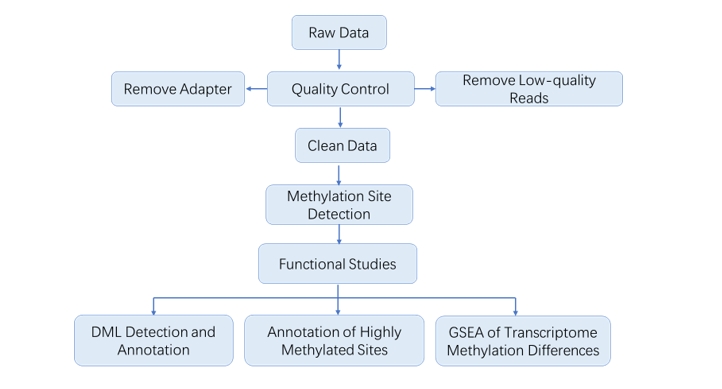
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Nanopore RNA Methylation Sequencing for your writing (customization)
Explore RNA methylation with CD Genomics' Nanopore RNA Methylation Sequencing. We offer direct RNA sequencing and comprehensive bioinformatics analysis to uncover epigenetic patterns. Contact us to advance your research insights.
Reference
- Jonkhout N, Tran J, Smith MA, et al. The RNA modification landscape in human disease. RNA. 2017, 23(12):1754-69.
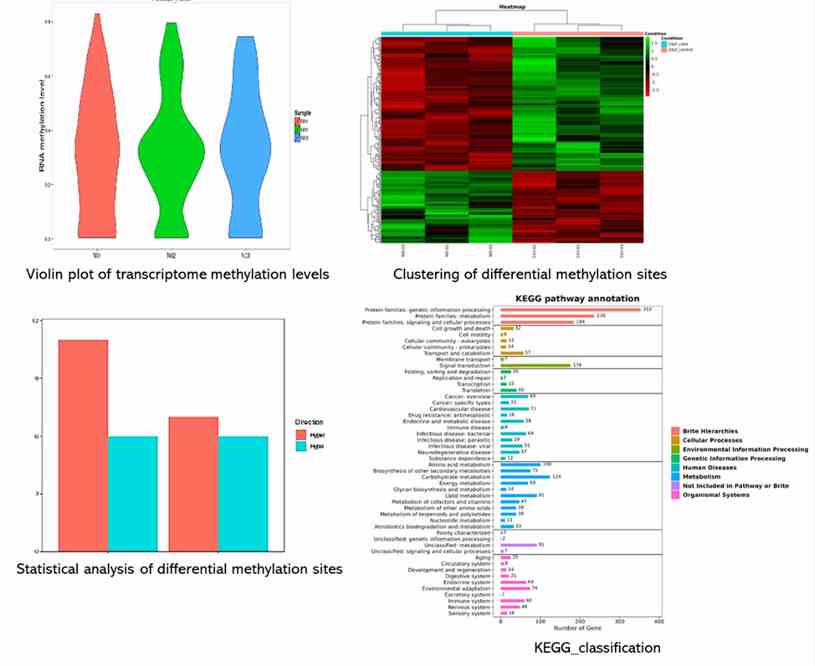
Partial results are shown below:
1. What molecules are involved in RNA modification processes?
In the process of RNA modification, several key molecules are involved. Writers, such as METL3 and METTL14, catalyze the methylation of RNA, with WTAP playing a crucial role in this methyltransferase complex. These enzymes facilitate m6A methylation of mRNA (and other nuclear RNAs) both in vitro and in vivo. On the other hand, erasers like FTO and ALKBH5 are responsible for removing m6A methylation signals from RNA, thus mediating the demethylation process. Readers, on the other hand, are proteins that interpret the modified information in RNA and participate in downstream processes like translation and splicing. Proteins with YTHDF domains, for example, can recognize and bind to m6A in mRNA, leading to a decrease in mRNA stability and promoting its degradation.
2. What types of RNA methylation can be detected with Nanopore RNA Methylation Sequencing?
This method can detect various RNA methylation modifications, including N6-methyladenosine (m6A), 5-methylcytosine (m5C), and others.
3. What research applications can benefit from Nanopore RNA Methylation Sequencing?
This technology is valuable for studying epigenetic regulation of gene expression, RNA processing, and various biological processes influenced by RNA modifications. It enables researchers to explore dynamic changes in RNA methylation patterns under different conditions.
4. How does Nanopore RNA Methylation Sequencing compare to other methods like MeRIP-seq or CM-seq?
Unlike antibody-based methods (e.g., MeRIP-seq) or chemical modification approaches (e.g., CM-seq), Nanopore RNA Methylation Sequencing directly reads RNA sequences and modifications in real-time, offering advantages in comprehensive coverage and reduced bias.
5. What are some typical outputs of Nanopore RNA Methylation Sequencing?
Outputs include detailed methylation profiles of RNA transcripts, visualization of methylation patterns (e.g., violin plots), differential methylation analysis, and functional annotations of methylated sites across the transcriptome.
FIONA1-Mediated m6A Modification Regulates the Floral Transition in Arabidopsis
Journal: Advanced Science
Impact factor: 17.521
Published: 05 January 2022
Background
m6A is a common mRNA modification critical for gene regulation, affecting mRNA processing, stability, and translation. Techniques like m6A-seq, m6A-CLIP, and nanopore sequencing enable precise mapping of m6A sites across the transcriptome. In Arabidopsis, m6A deposition involves complex methyltransferases targeting specific mRNA regions and motifs. FIO1, identified as a novel m6A methyltransferase, influences flowering time by regulating m6A levels in key genes like SOC1. Nanopore sequencing reveals insights into m6A dynamics, highlighting FIO1's role in plant development and gene expression control.
Materials & Methods
Sample Preparation
- Arabidopsis (A. thaliana)
- Six-day-old seedlings
- Plasmid construction
- Total RNA extraction
Sequencing
- Nanopore Direct RNA Sequencing
- GridION
- m6A-IP-qPCR
- RNA Immunoprecipitation
- m6A modification site analysis
- Differential expression analysis
- Alternative splicing analysis
- Expression analysis
- Statistical analysis
Results
Disrupting FIO1 in Arabidopsis reduces global mRNA m6A levels mildly. FIO1, similar to human METTL16, contains a conserved methyltransferase domain and is broadly expressed in Arabidopsis tissues. Mutants (fio1-1 and fio1-2) exhibit early flowering, with fio1-2 showing decreased FIO1 expression. Analysis confirms approximately 14% and 10% decreases in m6A levels in total RNA and mRNA of fio1-2 mutants compared to wild-type plants, indicating FIO1's involvement in m6A methylation.
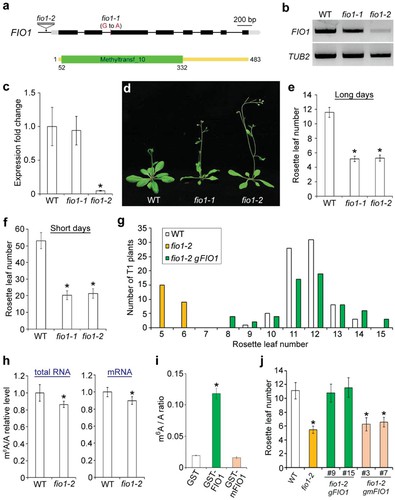 Figure 1 FIO1 affects flowering and mRNA m6A levels in Arabidopsis.
Figure 1 FIO1 affects flowering and mRNA m6A levels in Arabidopsis.
Nanopore direct RNA sequencing of fio1 mutants identified 3459 hypomethylated m6A sites in coding sequences (CDS). This approach, using xPore analysis, highlighted a preference for the YHm6AGA motif and revealed reductions in m6A levels mainly before stop codons. Functional analysis linked these changes to early flowering in fio1 mutants, confirming FIO1's role as an m6A methyltransferase essential for mRNA modification and developmental regulation in Arabidopsis.
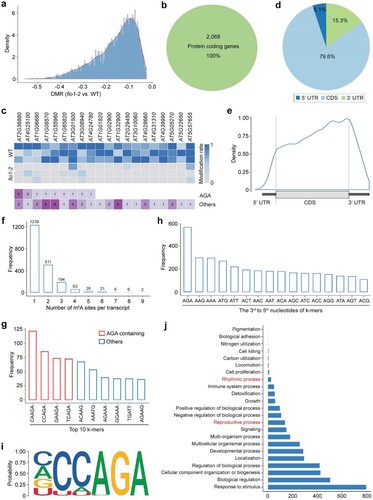 Figure 2 Distribution of hypomethylated sites in fio1-2.
Figure 2 Distribution of hypomethylated sites in fio1-2.
FIO1-mediated m6A methylation in Arabidopsis influences transcript abundance and alternative polyadenylation (APA), primarily affecting genes enriched in coding sequences (CDS). Hypomethylated sites in fio1 mutants correlate with downregulated gene expression, including the key flowering regulator SOC1. FIO1 directly modulates SOC1 and other circadian clock genes, lengthening their expression period and indirectly impacting flowering time. These findings highlight FIO1 as a distinct m6A methyltransferase targeting specific mRNA regions to regulate developmental processes in plants.
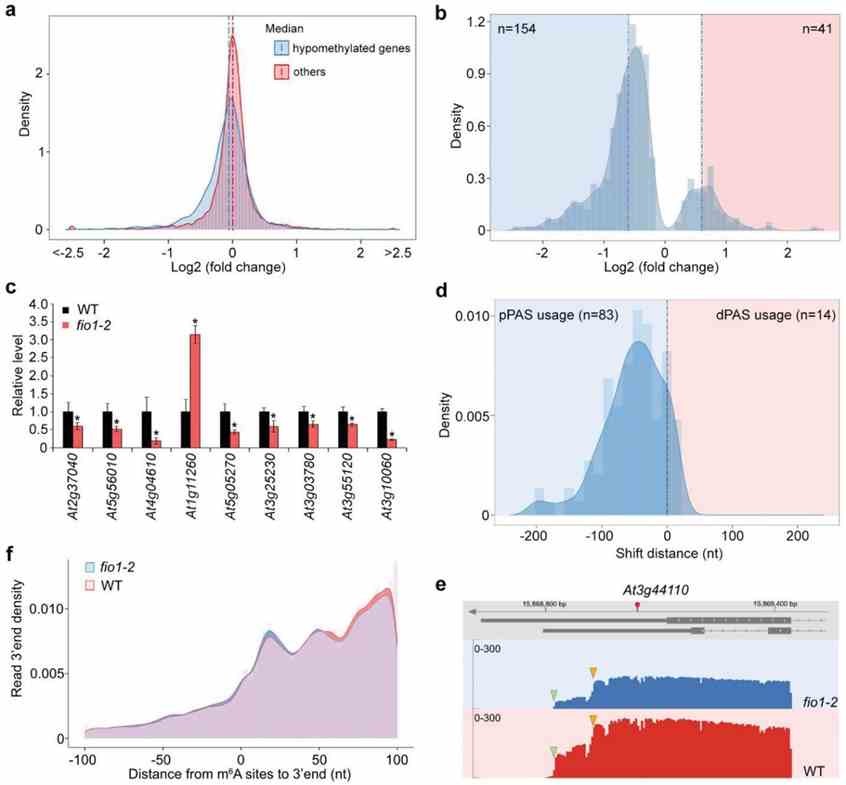 Figure 3 FIO1 regulates transcript abundance and alternative polyadenylation.
Figure 3 FIO1 regulates transcript abundance and alternative polyadenylation.
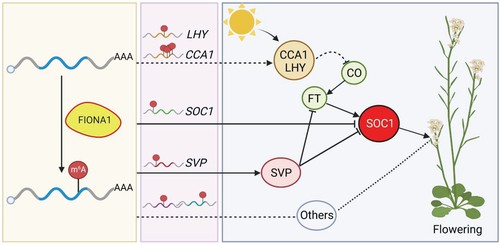 Figure 4 A proposed model depicting the function of FIO1 in m6A modification and flowering time control in Arabidopsis.
Figure 4 A proposed model depicting the function of FIO1 in m6A modification and flowering time control in Arabidopsis.
Conclusion
This study reveals FIO1 as a unique m6A methyltransferase in plants, primarily modifying coding sequences of specific protein-coding transcripts. FIO1-mediated m6A methylation influences transcript abundance and alternative polyadenylation, while showing minimal effect on alternative splicing. Importantly, FIO1 regulates SOC1 and its upstream regulators, thereby modulating flowering time. These findings enhance our understanding of plant m6A dynamics and draw parallels with METTL16-mediated RNA modification in animals.
Reference
- Xu T, Wu X, Wong CE, et al. FIONA1‐mediated m6A modification regulates the floral transition in Arabidopsis. Advanced Science. 2022, 9(6):2103628.
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


