
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics offers DAP-seq services using advanced genomic technologies to analyze transcription factor binding. Our high-throughput approach identifies TF-DNA interactions, providing insights into gene regulatory networks across various research areas. This service is ideal for unraveling gene expression mechanisms in both plants and animals, facilitating advancements in biological research.
The Introduction of DAP-Seq
Comprehensive identification of TFBS (transcription factor binding sites) in whole genome-wide is essential for characterizing regulatory elements and TF function. ChIP-Seq (Chromatin immunoprecipitation sequencing) is a powerful approach for determining in vivo TFBS. However, the method is limited in scale as it depends on specific antibodies for each transcription factor, which can be technically challenging and expensive. Currently, alternatives methods DAP-Seq can offer a simple and high-throughput method to examine TF binding to its cognate target (gDNA) in a chromatin-free context, while maintaining important information related to primary genome sequence and DNA methylation. The method does not require the specific antibodies or gene-specific primers, suitable for all eukaryotes, leading to a powerful tool for understanding regulatory elements and TF function.
The full name of DAP-seq stands for DNA affinity purification sequencing. This technique involves identifying transcription factor binding sites (TFBS) through the in vitro expression of transcription factors, without being constrained by antibodies or species, and possessing high-throughput advantages. Since its inception, this technology has been widely applied in the study of transcriptional regulation and epigenomics.
Principle of DAP-seq Experiment:
In DAP-seq experiments, the externally expressed protein and DNA are subject to affinity purification, followed by high-throughput sequencing of the DNA that bound to the protein. The fundamental process involves constructing the coding sequence (CDS) of the transcription factor into a vector containing an affinity tag, creating a protein expression vector and performing in vitro protein expression to produce a fusion protein of the transcription factor and the affinity tag. Subsequently, genomic DNA from the sample is extracted, a DNA library is constructed, and the in vitro expressed transcription factor with the affinity tag is combined with the DNA library. The bound DNA is then eluted and subjected to sequencing.
Comparison of DAP-seq and ChIP-seq Technologies
| Technology Name | DAP-seq | ChIP-seq |
|---|---|---|
| Experimental Mode | In vitro | In vivo |
| Requires Specific Antibodies | No | Yes |
| Suitable for Non-model Organisms | Yes | No |
| Time Cost | Low | High |
| High Throughput | Yes | No |
Advantages of DAP-Seq
- Suitable for high-throughput sample processing of entire TF ORF clone collections.
- Does not require the specific antibodies for each transcription factor, relatively cost-effective, and suitable for all eukaryotes.
- DAP-Seq retains many of the tissue/cell-line specific secondary modifications and features present in whole genome-wide.
- The enrichment process of DNA fragments at TF binding sites is simpler.
Applications of DAP-Seq
- Analysis and functional characterization of transcription factor binding sites
- Identification of histone modification types at specific DNA loci
- Study on the correlation between histone modifications and gene expression
DAP-Seq Workflow
Our highly experienced expert team executes quality management following every procedure to ensure comprehensive and accurate results. Our DAP-Seq workflow is outlined below, including library prep, protein expression, DNA affinity purification, sequencing, and bioinformatics analysis.
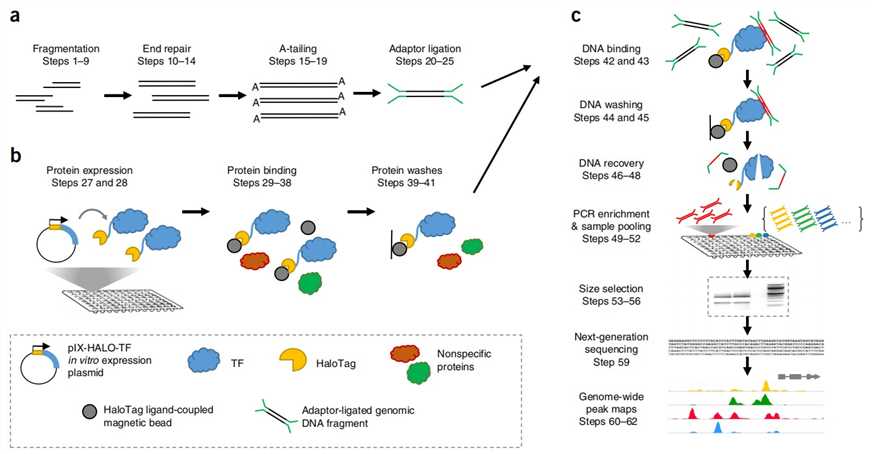 Fig 1. Experimental Procedure of DAP-seq. (Anna Bartlett et al., 2017)
Fig 1. Experimental Procedure of DAP-seq. (Anna Bartlett et al., 2017)
Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
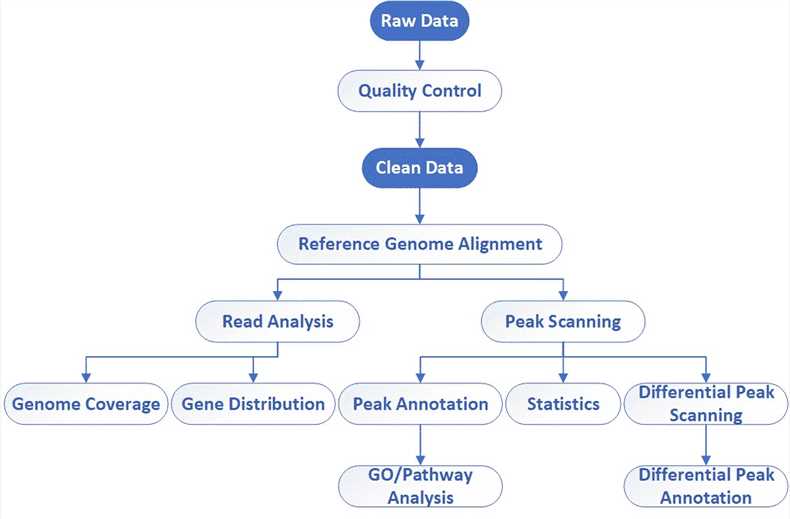
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in DAP-Seq for your writing (customization)
At CD Genomics, we provide you with high-quality sequencing and comprehensive bioinformatics analysis for your DAP-Seq project, enabling TFBS identification (transcription factor binding sites) in whole genome-wide. If you have additional requirements or questions, please feel free to contact us.
References
- Ronan C. O'Malley, S. shan C. Huang, L. Song, et al. Cistrome and epicistrome features shape the regulatory DNA landscape, Cell, 165 (2016) 1280–1292.
- Anna Bartlett, Ronan C. O'Malley, S. shan C. Huang, et al. Mapping genome-wide transcription-factor binding sites using DAP-Seq, Nature Protocols, 12 (2017) 1659–1672.
Partial results are shown below:
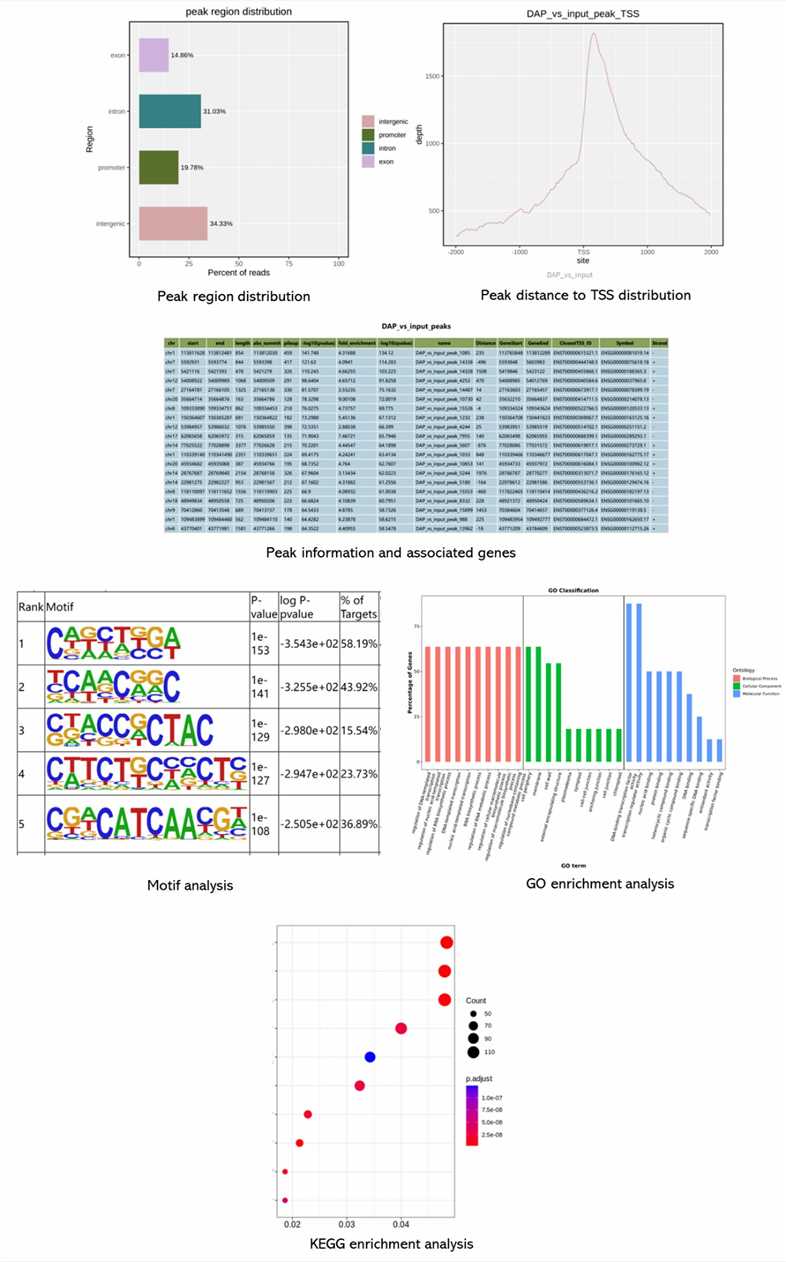
1. How does DAP-seq simplify the enrichment of TF binding sites?
DAP-seq simplifies enrichment by tagging TFs with a Halo-tag and using magnetic beads coated with ligands specific to the tag. This magnetic pull-down strategy replaces traditional immunoprecipitation, reducing experimental complexity.
2. Can DAP-seq detect DNA binding sites of regulatory factors?
Some regulatory factors bind to DNA indirectly through transcription factors, and DAP-seq can only detect binding sites of transcription factors that directly bind to DNA. Therefore, it is not recommended to use DAP-seq to detect binding sites of regulatory factors that bind to DNA indirectly.
3. Are there any limitations to DAP-seq?
Despite its advantages, DAP-seq relies on in vitro protein expression, which may introduce some differences compared to fully in vivo experiments.
4. How does DAP-seq differ from in vivo experiments?
In vitro conditions may not support proper protein folding or complex formation necessary for TF-DNA binding, impacting the functionality of some TFs.
A brassinosteroid transcriptional regulatory network participates in regulating fiber elongation in cotton
Journal: Plant Physiology
Impact factor: 7.479
Published: 21 December 2022
Background
Brassinosteroids (BRs) are essential substances found in rapeseed, regulating various developmental and physiological processes across the plant kingdom. BR deficiency or insensitivity leads to significant morphological changes. In cotton, BRs play a crucial role in fiber elongation, affecting gene expression related to cell wall biosynthesis and cytoskeleton dynamics. The transcription factor GhBES1.4 positively regulates cotton fiber elongation by binding to specific target genes, as identified through DAP-seq and RNA-seq analyses.
Materials & Methods
Sample Preparation
- Cotton
- Tissue samples
- gDNA extraction
- RNA extraction
Sequencing
- Library construction
- RNA-seq
- DAP-seq
- Transcriptional level analysis of differential genes
- Identification of target genes
- GWAS analyses
- Statistical analysis
Results
In this study, the authors utilized DAP-seq to identify 9,894 high-confidence binding regions of GhBES1.4 in cotton, predominantly located near transcription start sites. They further characterized GhBES1.4 binding motifs, revealing a preference for E-box elements and also binding to BRRE and novel motifs (MEME-3/4/5). Yeast one-hybrid and BLI assays confirmed GhBES1.4's affinity for these motifs, highlighting its role in regulating gene expression, including GhCPD, GhCYP90D1, and GhBRL, crucial for cotton fiber development.
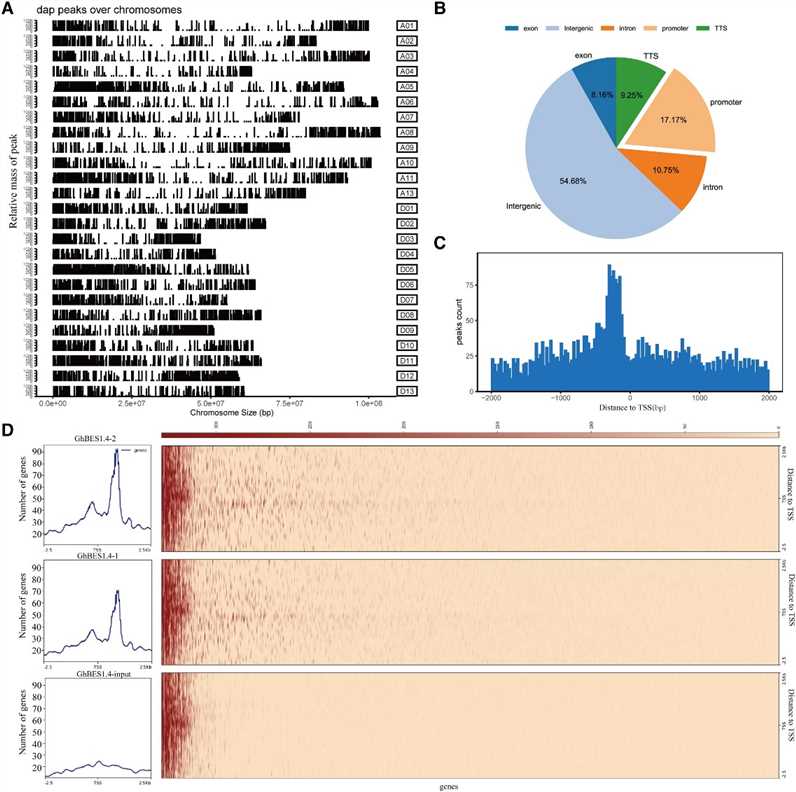 Figure 1 Identification of direct target genes of GhBES1.4 using DAP-seq.
Figure 1 Identification of direct target genes of GhBES1.4 using DAP-seq.
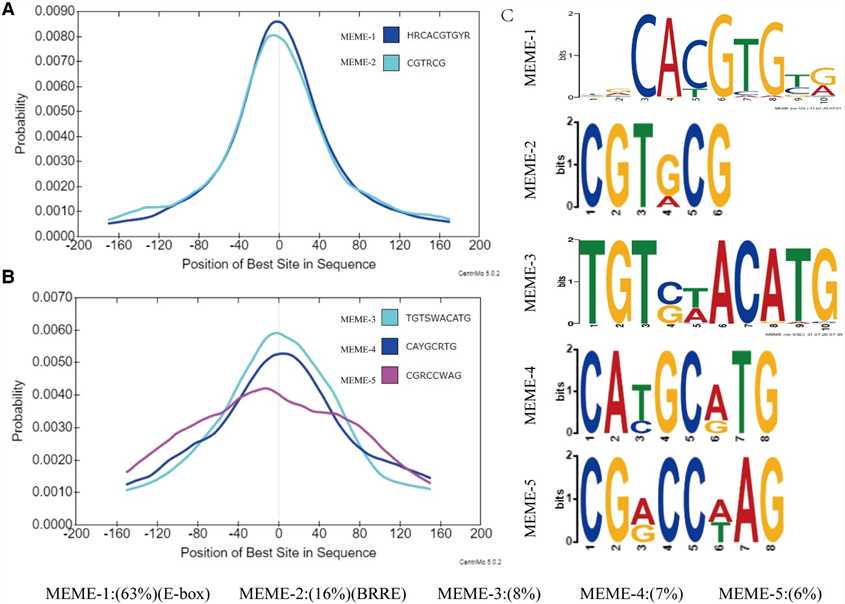 Figure 2 Characterization of the target site for GhBES1.4.
Figure 2 Characterization of the target site for GhBES1.4.
The authors generated GhBES1.4-OE and RNAi transgenic cotton plants, finding that GhBES1.4 positively regulates fiber elongation. GhBES1.4-OE plants showed increased fiber length and downregulated BR-related genes, while RNAi plants exhibited the opposite. RNA-seq analysis revealed 1,788 DEGs in GhBES1.4-OE and 1,566 in RNAi plants, involving genes related to cell wall elongation, primary wall synthesis, and tubulin assembly, indicating GhBES1.4's role in regulating fiber elongation-related gene expression.
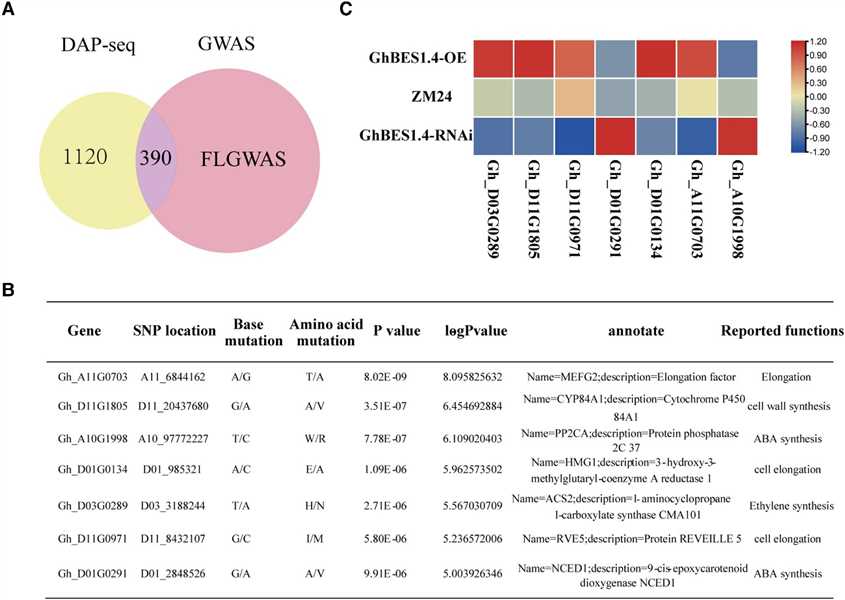 Figure 3 The integration of DAP-seq, RNA-seq, and genome-wide association study (GWAS) identifies the target genes for GhBES1.4 to regulate fiber elongation.
Figure 3 The integration of DAP-seq, RNA-seq, and genome-wide association study (GWAS) identifies the target genes for GhBES1.4 to regulate fiber elongation.
This study integrated DAP-seq, RNA-seq, and GWAS to identify GhBES1.4 target genes regulating cotton fiber elongation. GWAS identified 390 genes with SNP loci, of which 35 were highly associated with fiber length (FL). Transcriptome analysis and qPCR validated differential expression of 7 genes in transgenic materials. Biofilm interference assays confirmed GhBES1.4 binding to candidate gene promoters, and LUC activity assays showed direct activation by GhBES1.4. These genes, including RVE5, HMG1, and CYP84A1, influence fiber development via pathways like cell elongation and cell wall synthesis. Silencing GhCYP84A1 and GhHMG1 significantly shortened fiber length, highlighting their importance in GhBES1.4-mediated regulation of cotton fiber elongation.
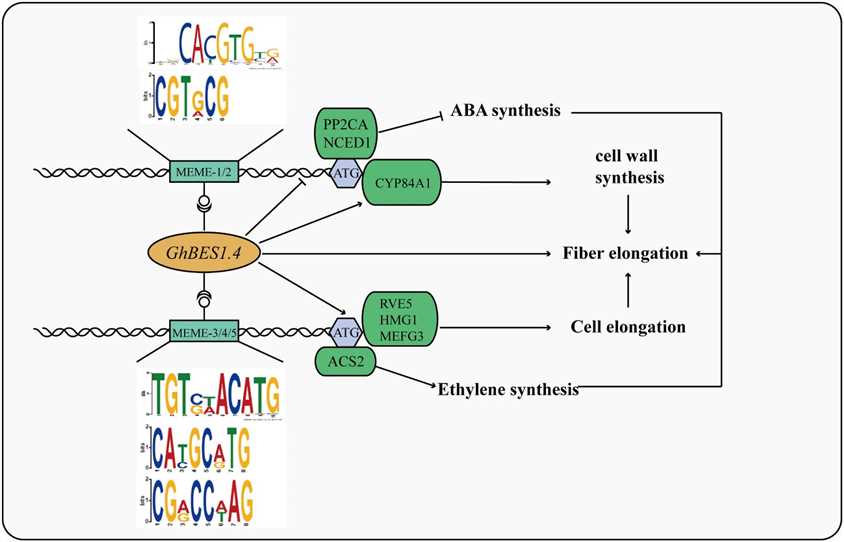 Figure 4 Mechanism of GhBES1.4 mediated fiber elongation.
Figure 4 Mechanism of GhBES1.4 mediated fiber elongation.
Conclusion
This study demonstrates the effectiveness of DAP-seq for identifying GhBES1.4 target genes in cotton, revealing 1,531 potential targets. This approach offers a rapid and robust method to unravel TF-mediated regulatory networks in plants like cotton.
Reference
- Liu L, Chen G, Li S, et al. A brassinosteroid transcriptional regulatory network participates in regulating fiber elongation in cotton. Plant Physiology. 2023, 191(3):1985-2000.
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


