
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics has further optimized the process to exceed PacBio's benchmarks in output and read length to support long-read metagenomic sequencing, and reduce some splicing errors and effectively improve the resolution of microbial community profiling.
The Introduction of Long-Read Metagenomic Sequencing
Metagenomics is defined as the direct genetic analysis of genomes contained within an environmental sample. The field initially started with the cloning of environmental DNA, followed by functional expression screening, and was then quickly complemented by sequencing of environmental DNA. Microbial samples are from nearly everywhere. Examples include microenvironments within and on the human body in healthy and diseased states, soil samples, plant roots, marine environments, wastewater treatment plants, urban areas, and so on.
In metagenomic experiments, total genomic DNA isolated from the environment are sequenced. It allows the researcher to identify the genes possessed and the metabolic processes performed by those microbial communities including unculturable organisms in the environment.
While many natural microorganisms cannot be isolated and cultured or cloned, some microbial biodiversity has been missed in sample preparation and sequencing. Now, CD Genomics provides long-read metagenomics sequencing service on PacBio SMRT System in a culture-free method, which could sequence thousands of organisms in parallel, and reduce some splicing errors and effectively improve the resolution of microbial community identification. Long-read metagenomics sequencing also helps researches generating new insights into the function and pathway facets of the microbiome, trying to understand and elucidate the relations between microbes and its habitat/host.
The data analysis is based on the highest quality single-molecule CCS reads with no need of assembly for species-level classification, functional insight and pathway enrichment study, could restore the information of microbial community in the environment. Long-read metagenomics sequencing is a powerful way to obtain microbial genomes with the highest accuracy and reliability.
Key Features and Advantages of Long-Read Metagenomic Sequencing
- Longest average read lengths, with~50% of reads longer than 50kb, which exceeds the size of repetitive elements in the average bacterial genome.
- No DNA amplification.
- Highest consensus accuracy, low sequencing-context bias
- Novel bioinformatics analysis programs and pipelines
- Well-experienced personnel
Applications of Long-Read Metagenomic Sequencing
Environmental Microbiology:
- Study microbial diversity and ecological functions in soil, water, and other environmental samples.
- Explore microbial community roles and interactions within ecosystems.
Human Microbiome Research:
- Investigate microbial composition and functions across human body sites (e.g., gut, oral cavity, skin).
- Examine relationships between microbiota and health or disease states, such as metabolic syndrome or inflammatory bowel diseases.
Industrial Microbiology:
- Optimize microbial community structures and functions in fermentation and bioprocessing.
- Develop and screen novel microbes and enzymes for industrial applications.
Agricultural Sciences:
- Study soil microbial communities' impact on crop growth and health.
- Develop microbial fertilizers and biocontrol agents to enhance agricultural productivity and sustainability.
Long-Read Metagenomic Sequencing Workflow
Our proficient team of experts implements rigorous quality management at each stage to ensure comprehensive and accurate results. Here is an overview of the typical workflow for Long-Read Metagenomic Sequencing:

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
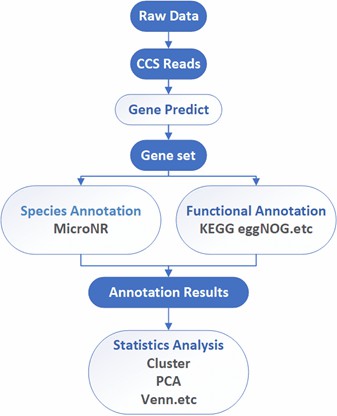
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Long-Read Metagenomic Sequencing for your writing (customization)
PacBio System is highly robust and cost-effective and should be the platform of choice in metagenomics sequencing, particularly for complex sample and low-diversity microbial communities. CD Genomics will be your best companion in long-read metagenomic sequencing. Please contact us for more information and a detailed quote.
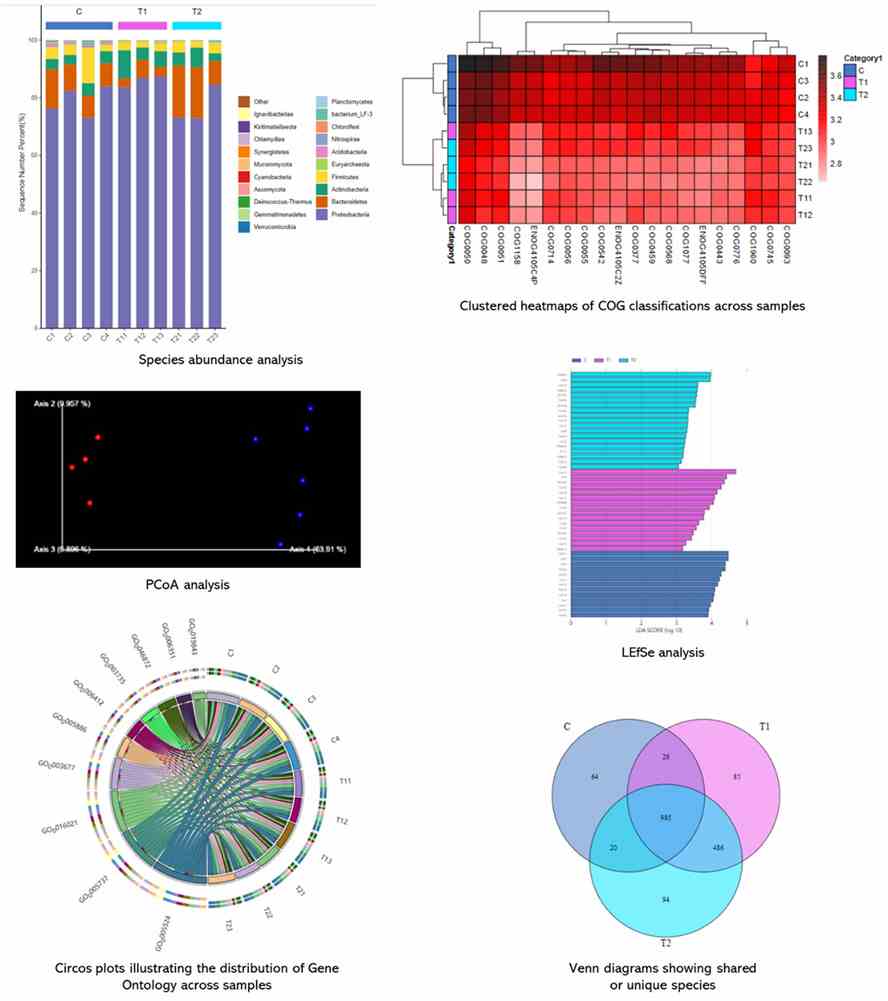
Partial results are shown below:
1. Why are longer sequencing reads preferable for metagenomics studies?
Longer sequencing reads offer several advantages for metagenomics studies, bolstering both the accuracy and comprehensiveness of genomic analyses:
- Enhanced Genome Assembly: The extended length of sequencing reads significantly improves the resolution of complex genomic regions, such as repetitive sequences and structural variations within microbial genomes. This leads to more contiguous and accurate genome assemblies when compared to the assemblies derived from shorter reads.
- Full-Length Gene and Operon Capture: The increased read length enhances the probability of capturing full-length genes and operons within a single read. This facilitates more precise functional annotations and pathway analyses, enabling a better understanding of microbial functions and interactions.
- Improved Resolution of Microbial Diversity: By spanning multiple variable regions within microbial genomes, longer reads allow for more precise identification and classification of microbial species and strains within complex communities. This improved resolution is crucial for studying microbial diversity and ecosystem dynamics.
- Reduction of Chimeric Assemblies: Short reads can often result in chimeric assemblies, where sequences from different genomes are incorrectly combined. The greater sequence context provided by longer reads mitigates this issue, reducing assembly errors and enhancing the overall accuracy of the genomic data.
- Insights into Microbial Genomic Structure: Longer reads provide valuable insights into the genomic structures of microbes, including gene clusters, mobile genetic elements, and genomic rearrangements. These insights are essential for understanding microbial evolution, adaptation, and the mechanisms underpinning microbial functions in various environments.
2. How does Long-Read Metagenomic Sequencing handle complex microbial communities?
Long-read sequencing technology can span repetitive regions and complex genomic structures more effectively than short-read technologies. This capability reduces assembly errors and improves the accuracy of microbial genome reconstructions within diverse microbial communities.
3. How does long-read sequencing differ from short-read sequencing?
Long-read sequencing generates reads spanning thousands to tens of thousands of base pairs, in contrast to short-read sequencing, which produces reads typically between 50 and 300 base pairs in length. The extended read lengths inherent in long-read sequencing confer a distinct advantage in elucidating complex genomic architectures, repetitive sequences, and complete transcript structures.
4. How does long-read sequencing improve data quality?
Long-read sequencing uses native DNA or RNA molecules, avoiding biases or errors introduced during synthetic amplification. It can also overcome assembly errors common in short-read sequencing, particularly in regions with repetitive sequences, thus enhancing data accuracy and completeness.
5. What are the challenges of long-read sequencing?
Despite its advantages, long-read sequencing faces challenges such as high costs, complex data analysis requirements, and technical demands for sample preparation. Some studies may require complementary technologies to obtain comprehensive genomic or transcriptomic information.
Uncovering microbiomes of the rice phyllosphere using long-read metagenomic sequencing
Journal: Communications Biology
Impact factor: 6.548
Published: 27 March 2024
Background
The phyllosphere harbors diverse microbial communities known as the plant microbiome, crucial for plant growth and health through nutrient uptake and disease resistance. Influenced by host genetics, defense mechanisms, environmental factors, and human activities, the microbiome's complexity impacts plant health unpredictably. Next-generation sequencing reveals microbial diversity and functional potential, yet faces biases. Long-read metagenomics overcomes limitations, enabling comprehensive genome reconstruction and identification of novel genetic elements like plasmids and bacteriophages. Applied to rice microbiomes, it unveiled new insights, highlighting its potential for plant microbiome research.
Materials & Methods
Sample Preparation
- Rice plants
- Bacterial cell enrichment
- DNA extraction
Sequencing
- 16S rRNA sequencing
- SMRTbell libraries construction
- Long-read metagenomic sequencing
- Assembly
- Gene annotation
- Classification of circular contigs
- Microbial composition analysis
- Predicted gene function
- Statistical analysis
Results
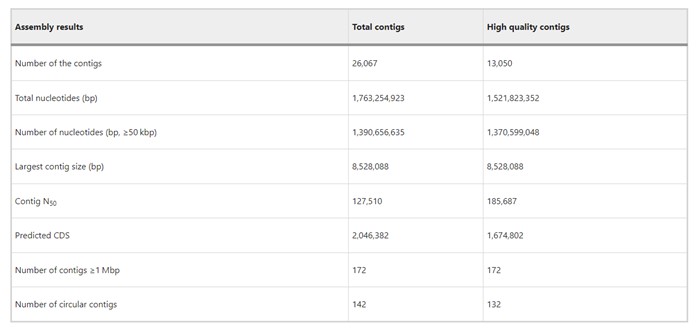
The authors used PacBio Sequel II to sequence leaf-associated microbial DNA, generating 140 Gbp data with an average read length of 17 kbp and mean library size of 15 kbp. They assembled 26,067 contigs (N50 = 128 kbp), including 142 circular contigs. Over half were high-quality (>5 read depths), representing 80% of reads and >90% of nucleotides in contigs ≥50 kbp. All contigs ≥1 Mbp were high-quality, confirming reliability for studying bacterial community composition and functions in the rice phyllosphere microbiome.
Table 1. Summary of assembly results
The study utilized long-read metagenomics to analyze microbial composition using 16S rRNA genes. They identified 669 16S rRNA genes on 561 contigs, with many representing potentially novel bacterial species. Taxonomic analysis revealed 463 sequences with ≥97% identity to known taxa, identifying 59 bacterial species. Comparison with full-length 16S rRNA amplicon and short-read sequencing data highlighted higher Actinobacteria abundance in the long-read metagenome, emphasizing its accuracy in identifying microbial communities in rice phyllosphere.
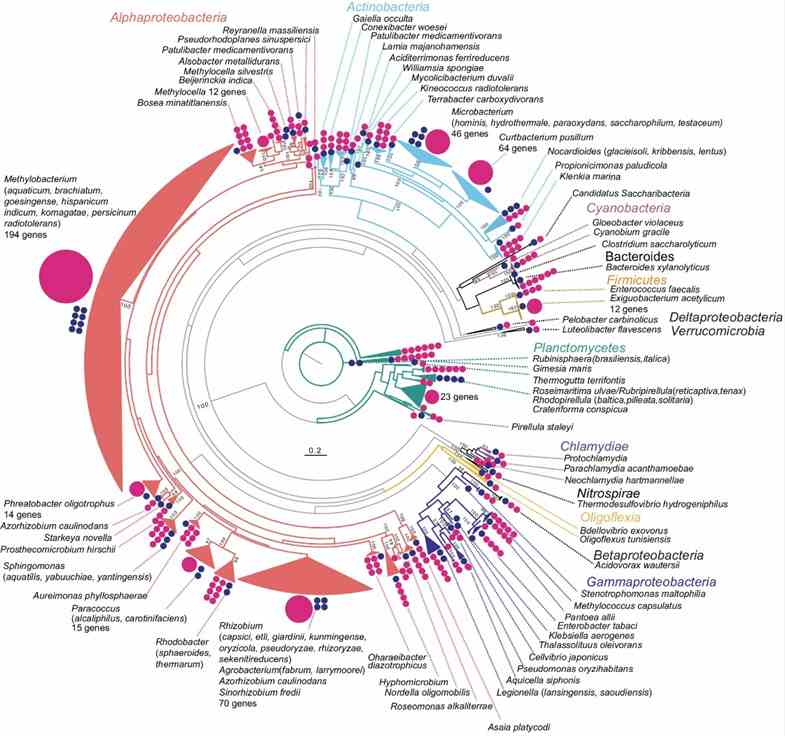 Fig. 1: Overview of the phylogeny of 16S rRNA genes detected in the metagenome.
Fig. 1: Overview of the phylogeny of 16S rRNA genes detected in the metagenome.
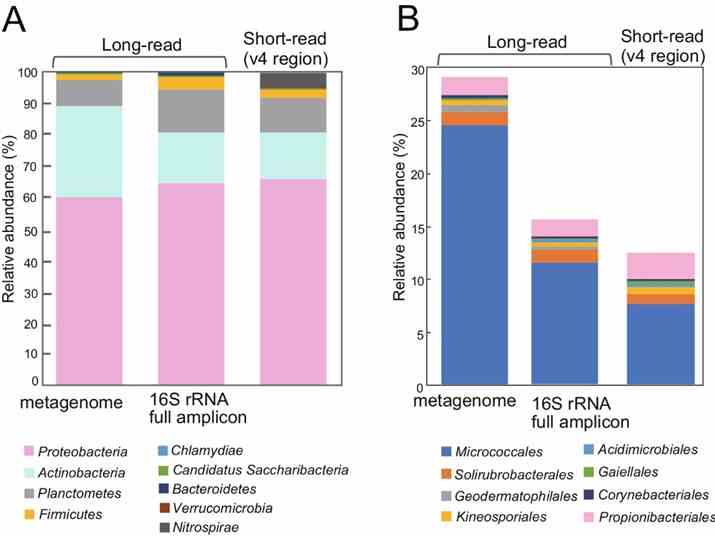 Fig. 2: Relative abundance of 16S rRNA genes detected in the metagenome, compared to 16S rRNA gene full-length amplicon sequences and short reads.
Fig. 2: Relative abundance of 16S rRNA genes detected in the metagenome, compared to 16S rRNA gene full-length amplicon sequences and short reads.
The study identified 2,046,382 predicted genes, with 364,262 annotated using the COG database. Methylobacterium dominated functional categories like amino acid metabolism and carbohydrate transport. They reconstructed 142 circular contigs, including bacterial chromosomes and a megaplasmid. Novel species were found, and diverse plasmids, bacteriophages, and VirB/VirD4 T4SS-related elements were characterized, revealing extensive microbial diversity and genetic potential in the rice phyllosphere using long-read metagenomics.
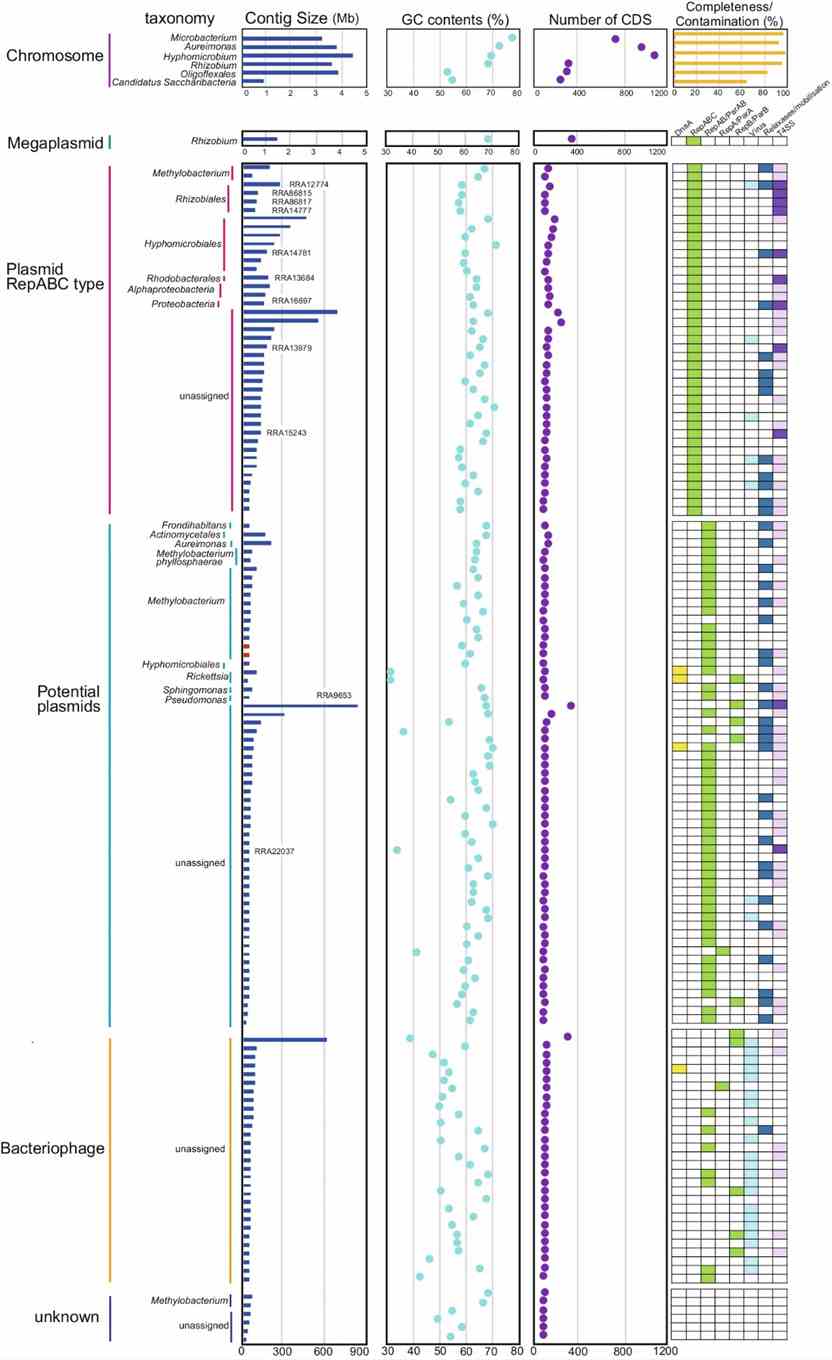 Fig. 3: Characteristics of circular contigs (n = 142).
Fig. 3: Characteristics of circular contigs (n = 142).
Long-read metagenomics revealed the complete genome of Candidatus Saccharibacteria bacterium RRA8490, phylogenetically related to human oral microflora. RRA8490 differed significantly from known strains (52.2–54.2% amino acid identity), lacking amino acid and fatty acid synthesis genes but encoding glucose metabolism pathways, ATP production mechanisms, type IV pili, and the cytochrome oxidase complex (CyoABCDE) for adaptation.
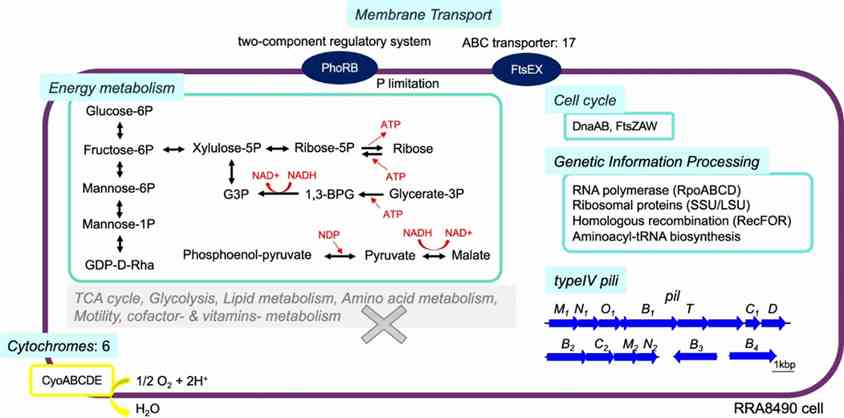 Fig. 4: Predicted metabolism of RRA8490, a potential new strain in the Candidatus Saccharibacteria phylum.
Fig. 4: Predicted metabolism of RRA8490, a potential new strain in the Candidatus Saccharibacteria phylum.
Conclusion
This study used enzymatic DNA extraction and long-read metagenomic sequencing to profile plant microbiota comprehensively. The method revealed diverse plasmids and bacteriophages, and characterized a Candidatus Saccharibacteria genome adapted to the rice phyllosphere. These findings underscore the utility of long-read sequencing for understanding plant-associated microbial ecology.
Reference
- Masuda S, Gan P, Kiguchi Y, et al. Uncovering microbiomes of the rice phyllosphere using long-read metagenomic sequencing. Communications Biology, 2024, 7(1): 357.
Here are some publications that have been successfully published using our services or other related services:
Bacterial communities of Cassiopea in the Florida Keys share major bacterial taxa with coral microbiomes
Journal: bioRxiv
Year: 2024
Production of a Bacteriocin Like Protein PEG 446 from Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotics and Antimicrobial Proteins
Year: 2024
Untangling the Role of Pathobionts from Bacteroides Species in Inflammatory Bowel Diseases
Journal: bioRxiv
Year: 2023
A chromosome-level genome resource for studying virulence mechanisms and evolution of the coffee rust pathogen Hemileia vastatrix
Journal: bioRxiv
Year: 2022
Streptomyces buecherae sp. nov., an actinomycete isolated from multiple bat species
Journal: Antonie Van Leeuwenhoek
Year: 2020
See more articles published by our clients.


