
 Sample Submission Guidelines
Sample Submission Guidelines
The Introduction of Nanopore
The complexity of short-read second-generation sequencing data assembly and the inability to reliably resolve repeat sequences or large genomic rearrangements were overcome by the third generation of sequencing, they all generate very long reads (1–100kb), which is distinct from next generation sequencing.
In 2014, Oxford Nanopore Technologies released nanopore sequencing in the form of the MinION, a handheld sequencer that uses a grid of membrane-embedded biological nanopores. The membrane in which the nanopores sit provides separation of two ionic solutions allowing an electrical current to flow through the nanopores. Long DNA molecules are prepared by adding a hairpin adaptor to one end of the double-stranded molecule before a helicase, and motor protein attached to the template unwind and thread single-stranded DNA through the nanopore channel. The DNA can be ratcheted through the pore one base at a time, with the nucleotide bases inducing characteristic changes in the electrical current running through the nanopore that are translated to base calls. As the sequencing process uses very few depletable reagents, the run can effectively continue until a satisfactory result is achieved.
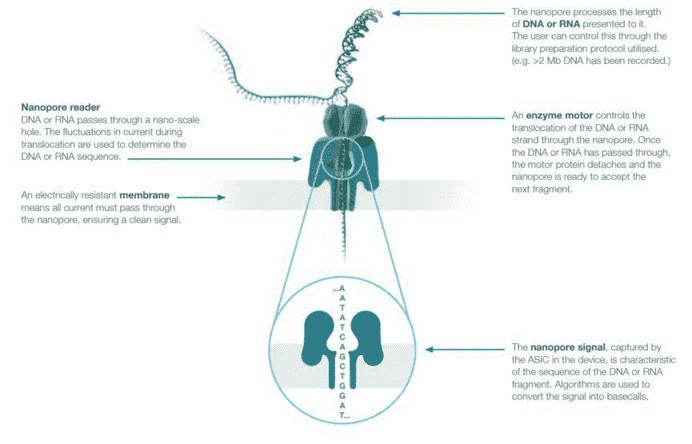 Figure 1. Nanopore sequencing overview.
Figure 1. Nanopore sequencing overview.
Advantages of Nanopore Full-Length Transcripts Sequencing
- The slightly longer mappable length (> 2 Mb).
- ONT MinION provides very high throughput as the nanopores can sequence multiple molecules.
- The cost for ONT data generation is lower.
- Precise quantification of isoforms: Accurately differentiating and identifying isoforms and new transcripts; a correlation of 98.9% is reported between the quantitative assessements and theoretical values.
- Absence of GC and PCR bias: Since the library preparation process does not include a PCR step, PCR bias is eliminated; furthermore, there is no GC bias.
- Wide range of transcript detection: Transcripts, usually spanning a few thousand nucleotides, are detectable without fragment selection, allowing for identification of transcripts from tens to tens of thousands of nucleotides.
- Direct detection of structural variants: Direct discovery of variable splicing and fusion genes is made possible.
Since sequencing cost is a significant obstacle of TGS application, the relatively high throughput and affordability makes ONT promising for many applications.
Application of Nanopore Full-Length Transcripts Sequencing
- Embryonic Development
- Cell Differentiation
- Cell Proliferation
- Neuronal Activity
- Immune Response
- Tumor Formation and Metastasis
Nanopore full-length transcriptome sequencing offers diverse applications across various biological research fields, including:
- Identification of Novel Transcripts: This technology aids in discovering new transcripts and splice variants without requiring a reference genome.
- Gene Structure Analysis: It enables precise examination of gene structures, including exon-intron boundaries, untranslated regions (UTRs), and polyadenylated tails.
- Detection of Fusion Genes and Alternative Splicing: Full-length transcriptome sequencing can directly identify fusion genes and splice variants, particularly beneficial for cancer research.
- Eukaryotic Transcript Analysis: The technology selectively enriches eukaryotic transcripts with poly-A tails during library preparation, enhancing their study.
- Study of lncRNAs: While primarily enhancing mRNA and poly-A tailed lncRNAs, it also provides insights into lncRNA structure and function. However, lncRNAs lacking poly-A tails cannot be enriched and remain undetected.
Nanopore Full-Length Transcripts Sequencing Workflow
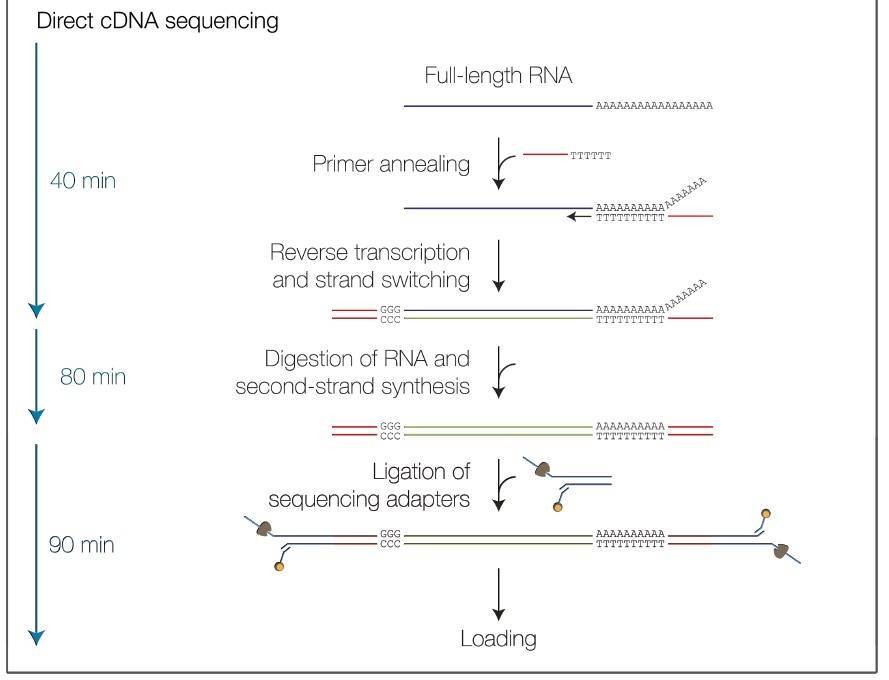
Service Specification
Sample Requirements
|
|
 |
Sequencing Strategy
|
 |
Bioinformatics Analysis
|
Analysis Pipeline
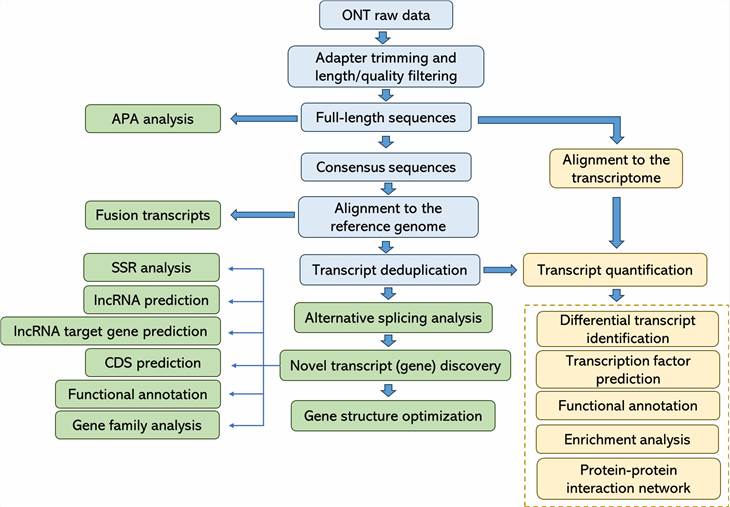
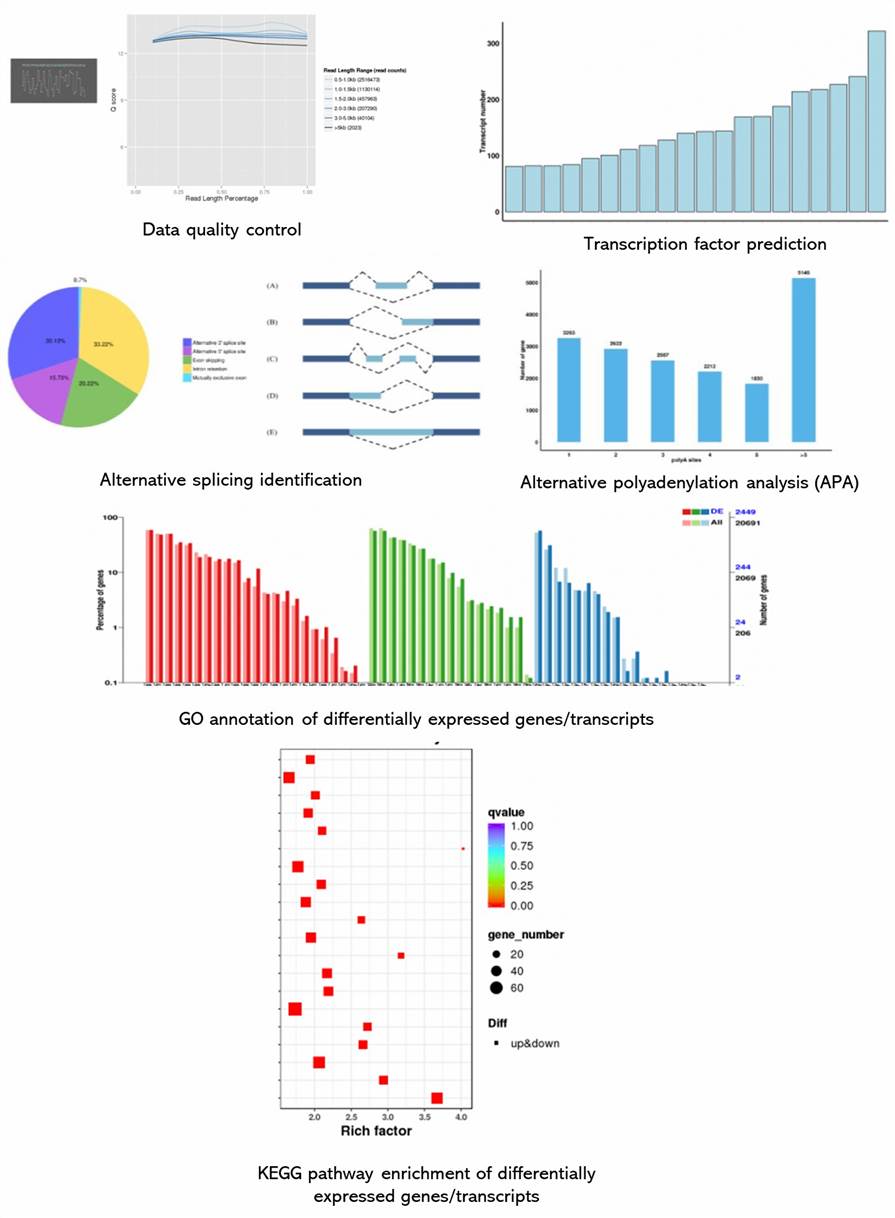
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Nanopore Full-Length Transcripts Sequencing for your writing (customization)
Reference:
- Weirather J L, Cesare M D, Wang Y, et al. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000research, 2017, 6:100.
1. Does full-length transcriptome sequencing require fragmentation and assembly?
Full-length transcriptome sequencing, based on PacBio or Nanopore third-generation sequencing platforms, does not require fragmentation and assembly. It directly generates complete transcripts containing 5' and 3' UTRs and poly-A tails. This approach enables precise analysis of alternative splicing and gene fusion events in species with a reference genome, addressing the challenges of shorter and incomplete transcript assemblies in species without a reference genome.
2. What is the accuracy of full-length transcriptome detection?
The base error rate of third-generation sequencing platforms is unbiased. For the Nanopore platform, using reference genome information for sequence correction can significantly enhance the accuracy of base correction.
3. Does full-length transcriptome sequencing capture all lncRNAs?
During the library construction process, RNA molecules with poly-A structures are identified, enriching mRNAs and some lncRNAs with poly-A tails from the total RNA. However, lncRNAs without poly-A structures cannot be enriched and sequenced in this manner, resulting in the detection of only a subset of lncRNAs.
4. How many biological replicates are generally recommended for Nanopore full-length transcriptome sequencing?
Research indicates that biological replicates improve the accuracy of identifying gene expression levels across the entire transcriptome, while increased sequencing depth mainly enhances the accuracy of detecting low-expression genes. It is recommended to have at least three biological replicates for each treatment condition. For studies involving samples with high biological variability or those aiming to detect subtle expression differences or fold changes, a higher number of biological replicates is needed. For example, clinical samples with significant individual differences may require 5-10 replicates per group, whereas cell line samples with lower biological variability may suffice with three replicates per group.
5. What are the differences between Nanopore full-length transcriptome sequencing and second-generation Illumina RNA sequencing?
The primary difference lies in the sequencing platforms. The Illumina platform typically uses paired-end 150 (PE150) sequencing, constructing small fragment libraries and performing sequencing by synthesis, which requires PCR amplification during library preparation and sequencing. It is mainly used for quantifying gene expression and differential expression analysis. In contrast, Nanopore full-length transcriptome sequencing does not require RNA fragmentation and can capture full-length transcripts from 5' to 3', providing comprehensive sequence and expression information. It has no bias towards fragment size and directly detects electrical signals, eliminating the need for synthesis during sequencing and resulting in a much lower GC bias compared to second-generation platforms. Additionally, because it does not require transcript assembly, it has significant advantages in detecting transcript-level structural variations such as alternative splicing, gene fusions, APA (alternative polyadenylation), and novel gene prediction.
Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns
Journal: Nature communications
Impact factor: 14.919
Published: 18 March 2020
Background
In various cancer types, mutations in the splicing factor SF3B1 have been associated with specific changes in splicing patterns, affecting conditions such as chronic lymphocytic leukemia (CLL), uveal melanoma, breast cancer, and myelodysplastic syndromes. SF3B1, a fundamental component of the spliceosome, interacts with pre-mRNA and exerts crucial control over the splicing process. Particularly noteworthy are mutations in SF3B1, notably the K700E mutation, which have been correlated with adverse clinical outcomes in CLL. While short-read sequencing has provided insights into splicing patterns influenced by SF3B1 mutations, long-read nanopore sequencing offers a more comprehensive perspective, unveiling novel details regarding splicing alterations. Nanopore sequencing facilitates the detection of full-length transcript molecules and enables precise evaluation of alternative splicing events, presenting promising avenues for cancer research and therapeutic interventions.
Methods
- Peripheral blood mononuclear cells
- RNA extraction
- Reverse transcribed
- Library preparation
- Oxford Nanopore sequencing
- PromethION sequencing
- Nanopore-sequencing statistics
- Spliced alignment and read correction
- Saturation analysis
- GO analysis
Results
In this pilot study, the authors used nanopore sequencing to analyze SF3B1K700E full-length transcripts in CLL samples. The authors found increased aberrant 3' splicing in SF3B1K700E, confirming previous findings. Additional samples were sequenced using nanopore technology, generating 257 million total reads. Despite RNA storage for 5 years, minimal degradation was observed. Strong gene expression correlation between minION and PromethION sequenced samples allowed data combination for subsequent analyses.
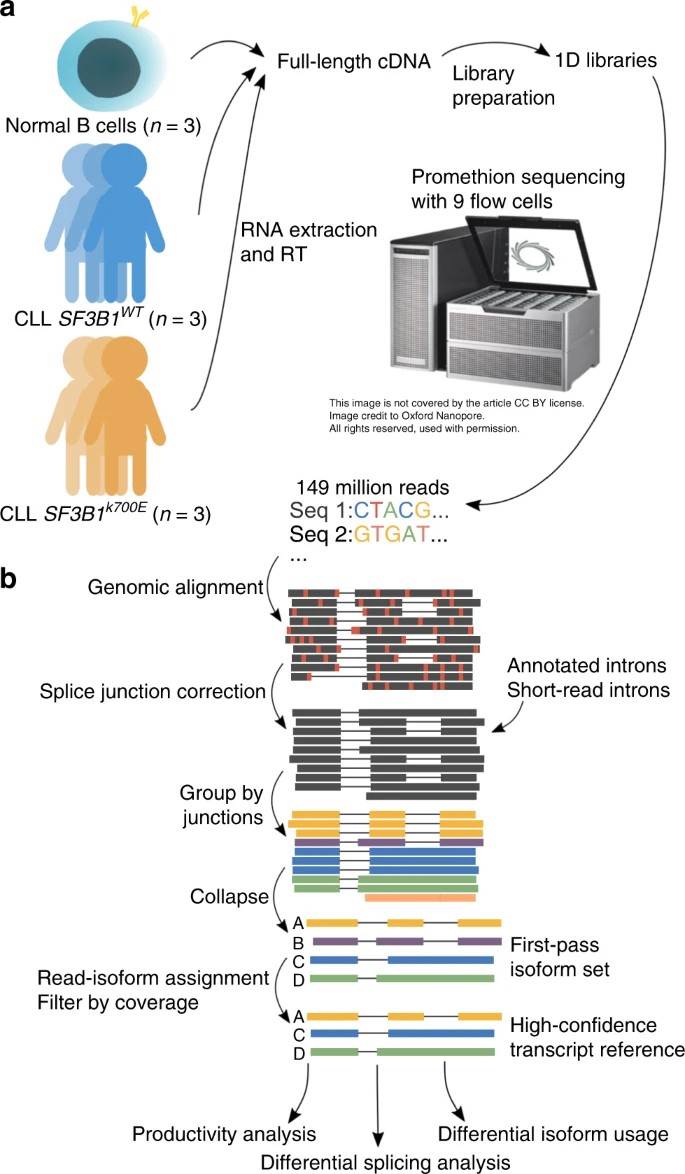 Fig. 1: Long-read nanopore sequencing and FLAIR analysis to identify full-length transcripts associated with SF3B1 mutation in chronic lymphocytic leukemia.
Fig. 1: Long-read nanopore sequencing and FLAIR analysis to identify full-length transcripts associated with SF3B1 mutation in chronic lymphocytic leukemia.
In this study, the researchers sought to confirm the augmentation of alternative 3' splice-site (3'SS) utilization induced by SF3B1K700E through nanopore sequencing data. A comparative analysis of 3' splicing events in a chronic lymphocytic leukemia (CLL) cohort, initially identified via short-read Illumina sequencing, was conducted alongside nanopore sequencing results. To accurately classify alternative splicing (AS) events in isoforms, the authors devised an AS event caller within FLAIR. This approach facilitated the identification of significant differences in alternative 3' and 5' splice sites between SF3B1K700E and SF3B1WT. Noteworthy observations included the identification of an adenine-rich region upstream of the canonical 3'SS, aligning with previous research findings.
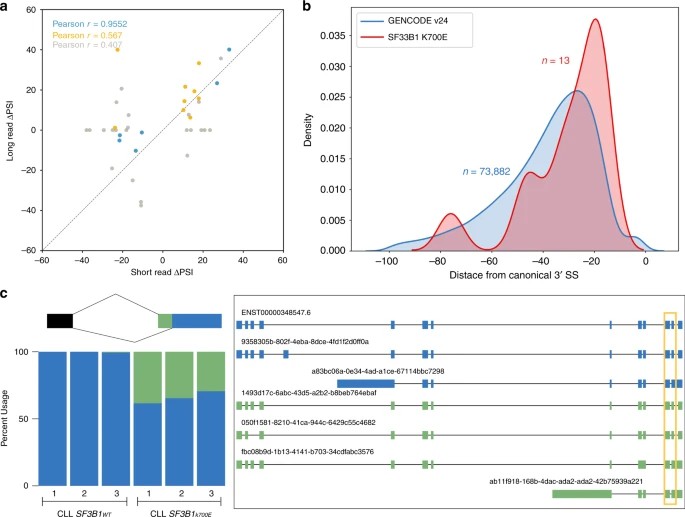 Fig. 2: Alternative 3'splicing patterns identified with nanopore-sequencing data are concordant with short read data and reveal additional splicing complexity.
Fig. 2: Alternative 3'splicing patterns identified with nanopore-sequencing data are concordant with short read data and reveal additional splicing complexity.
Short-read studies have linked mutant SF3B1 in CLL to an increase in transcripts with premature termination codons (PTCs). Full-length cDNA sequencing allows for more accurate detection of transcripts with PTCs and estimation of unproductive isoforms. Unproductive isoforms, defined by PTCs located 55 nucleotides or more upstream of the 3' most splice junction, may undergo nuclear retention or nonsense-mediated decay. In nanopore data, authors accurately identified known unproductive transcripts of SRSF1 and discovered additional unannotated isoforms, some of which are predicted to be unproductive.
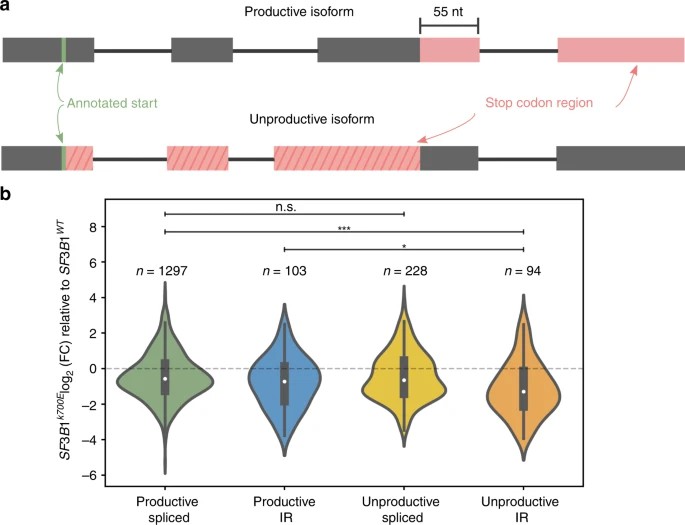 Fig. 3: Mutant SF3B1 downregulates unproductive, intron-retaining transcripts.
Fig. 3: Mutant SF3B1 downregulates unproductive, intron-retaining transcripts.
Conclusion
This study utilizes nanopore sequencing to examine full-length cDNA from CLL samples, revealing significant differential splicing events associated with SF3B1 mutation. The authors introduce FLAIR, a computational workflow for transcript analysis, and identify changes in 3'splice sites and downregulation of intron retention events. Full-length transcript analysis provides insights into alternative splicing and isoform abundance, highlighting the potential of nanopore sequencing for cancer and splicing research.
Reference:
- Tang, Alison D., et al. "Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns." Nature communications 11.1 (2020): 1438.
Here are some publications that have been successfully published using our services or other related services:
FIONA1-mediated methylation of the 3'UTR of FLC affects FLC transcript levels and flowering in Arabidopsis
Journal: PLoS Genetics
Year: 2022
The m6A writer FIONA1 methylates the 3'UTR of FLC and controls flowering in Arabidopsis
Journal: bioRxiv
Year: 2022
Complete Genome Sequence of the Lignocellulose-Degrading Actinomycete Streptomyces albus CAS922
Journal: Microbiology Resource Announcements
Year: 2020
See more articles published by our clients.


