
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics provides the accurate and cost-effective shallow whole genome sequencing service using Illumina or BGI DNBseq platforms to achieve genome-wide genetic variation for plants, animals and humans.
What is Shallow Whole Genome Sequencing
Shallow Whole Genome Sequencing (shallow WGS, also known as low pass whole genome sequencing) is a new and high-throughput technology to achieve genome-wide genetic variation accurately and cost-effectively with a broad range of species: cattle, pig, chicken, dog, cat, rat, mice, corn, rice, soybean and pea and humans. Based on shotgun sequencing, shallow WGS can be used to obtain whole genome sequence at a very low coverage (most frequently between 0.4x and 1x) with over 99% accurate variant calls. Shallow WGS returns more data, greater statistical power, and new rare variant discovery capabilities than traditional genotyping arrays and GBS method. Shallow WGS can be used in genome-wide association study (GWAS), evolutionary analysis, pharmacogenomics, molecular breeding, etc. In addition, Shallow WGS can be used to build a custom reference panel for a specific population.
Advantages of Shallow Whole Genome Sequencing
- More cost-effective than genotyping arrays and regular whole-genome sequencing
- More data, greater statistical power, and new rare variant discovery capabilities than traditional genotyping arrays
- Over 99% accurate variant calls across whole genome
- Flexible setup of new species or custom populations
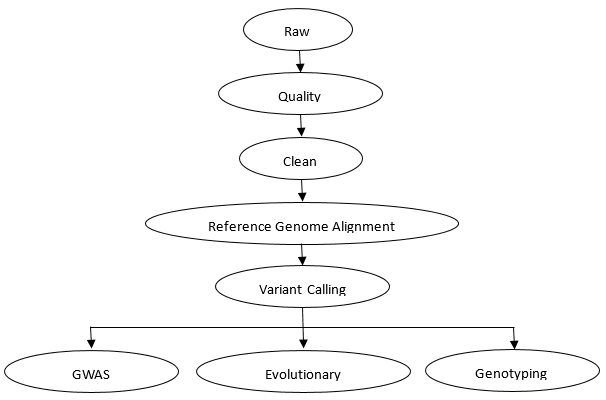
Shallow Whole Genome Sequencing Workflow
CD Genomics provides the accurate and cost-effective shallow whole genome sequencing and bioinformatics analysis for plants, animals and humans. Our professional expert team executes quality management, following every procedure to ensure confident and unbiased results. The general workflow for shallow whole genome sequencing is outlined below.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Data Analysis We provide customized bioinformatics analysis including:
|
Analysis Pipeline
CD Genomics provides full shallow whole genome sequencing service package including DNA extraction, library construction, sequencing, raw data quality control, and bioinformatics analysis. We can tailor this pipeline to your research interest. We can offer BAM and VCF data files for you. If you have additional requirements or questions, please feel free to contact us.
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Shallow Whole Genome Sequencing for your writing (customization)
1. How long does it take to get my results back?
Typically, 30-40 working days from sample qualified acceptance to data analysis report availability.
2. Which sequencing machines do you use for low pass whole genome sequencing?
Illumina or BGI DNBseq platforms can be used for sequencing.
3. What are the advantages of low pass whole genome sequencing over traditional genotyping arrays?
Low pass whole genome sequencing can be used to obtain whole genome sequence at a very low coverage (most frequently between 0.4x and 1x) with over 99% accurate variant calls. Over 10x more data than genotyping arrays at a similar or lower cost. Moreover, low pass whole genome sequencing allows to discover new rare variants. All these suggest that low pass whole genome sequencing outperforms genotyping arrays.
Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival
Journal: Cancers
Impact factor: 6.575
Published: 6 June 2020
Background
The FGFR family exhibits diverse genomic alterations, with FGFR1 amplification linked to poor prognosis and resistance to therapy in breast cancer. Clinical trials face challenges in obtaining fresh tissue biopsies for molecular analysis, leading to reliance on archival tissue with limited representation of tumor heterogeneity. Gene amplification evolves during cancer progression, emphasizing the need for real-time molecular profiling. Shallow whole-genome sequencing (sWGS) emerges as a cost-effective method for detecting circulating tumor DNA (ctDNA), aiding in treatment monitoring and prognostication. This study utilizes sWGS to explore ctDNA as a surrogate for FGFR1 amplification in metastatic breast cancer, correlating ctDNA levels with patient survival.
Methods
- Tissue and plasma samples
- Embedded in paraffin (FFPE)
- DNA extraction and Quantification
- Fluorescence In Situ Hybridization (FISH)
- Shallow Whole-Genome Sequencing sWGS (plasma-seq)
- Illumina MiSeq or NextSeq
- Statistical analysis
- Kaplan-Meier analysis
- Pearson correlation
Results
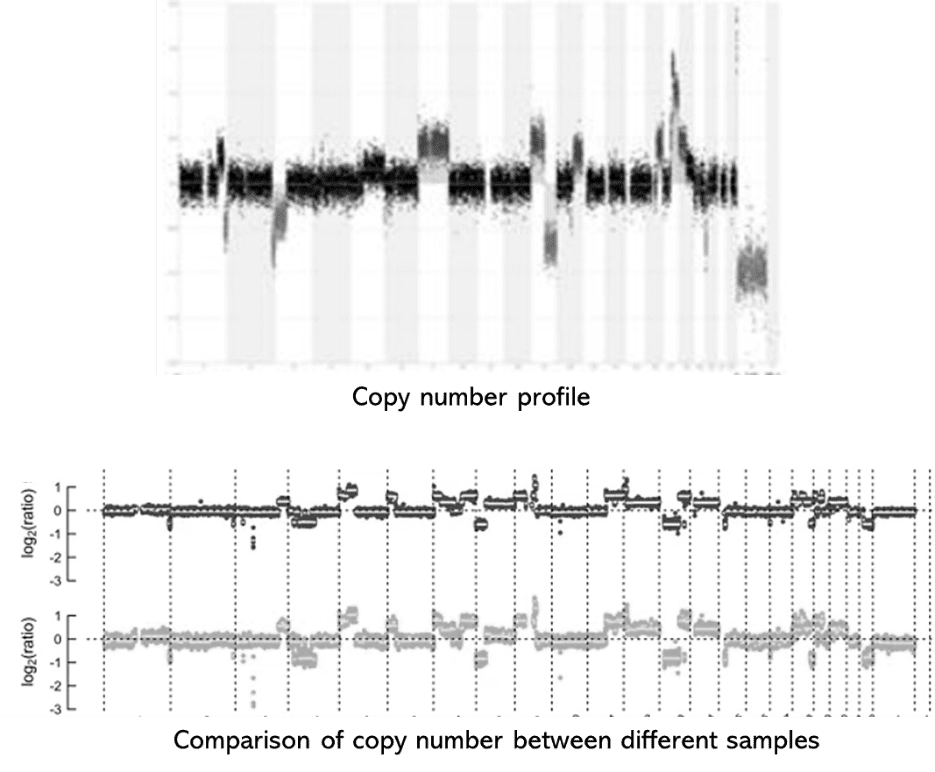
Twenty cases, including ten FGFR1-amplified and ten control cases, were analyzed for ctDNA using mFAST-SeqS, followed by sWGS irrespective of mFAST-SeqS results. Although mFAST-SeqS cannot detect FGFR1 amplification, sWGS identified amplifications down to 1%. A good correlation was observed between mFAST-SeqS z-scores and ichorCNA estimates. Cases with ctDNA allele fractions (AF) above 3% accurately identified FGFR1 amplification, but cases with low ctDNA AFs still detected amplification, highlighting the challenges in establishing detection thresholds. Correlation between log2ratio and tumor fraction was observed for amplified cases, with hypothetical copy numbers correlating with Oncoscan but not FISH results. The detection of FGFR1 amplification in plasma depends on both gene copy number and tumor fraction, making a generalized threshold challenging to establish.
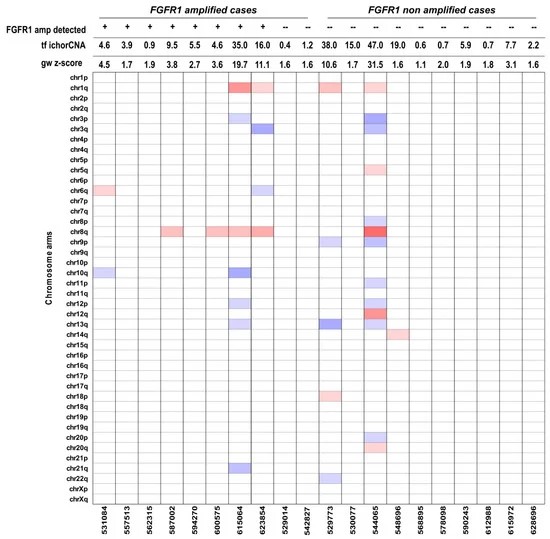 Fig 1. Heat-map of selected plasma samples with different values of genome-wide z-scores.
Fig 1. Heat-map of selected plasma samples with different values of genome-wide z-scores.
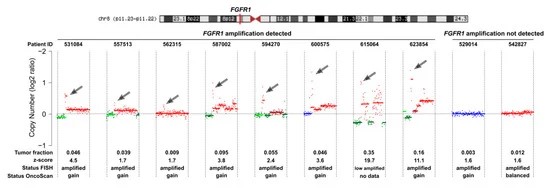 Fig 2. Copy number status of chromosome 8 of cell-free DNA (cfDNA) samples established with shallow whole-genome sequencing (sWGS).
Fig 2. Copy number status of chromosome 8 of cell-free DNA (cfDNA) samples established with shallow whole-genome sequencing (sWGS).
This study evaluated tumor fraction using ichorCNA with ULP-WGS in metastatic breast cancer. Stratification based on median genome-wide z-score (>2.2) and tumor fraction (>4.6) revealed significantly reduced overall survival (OS). Conversely, total cfDNA quantity showed no discernible relevance in OS discrimination.
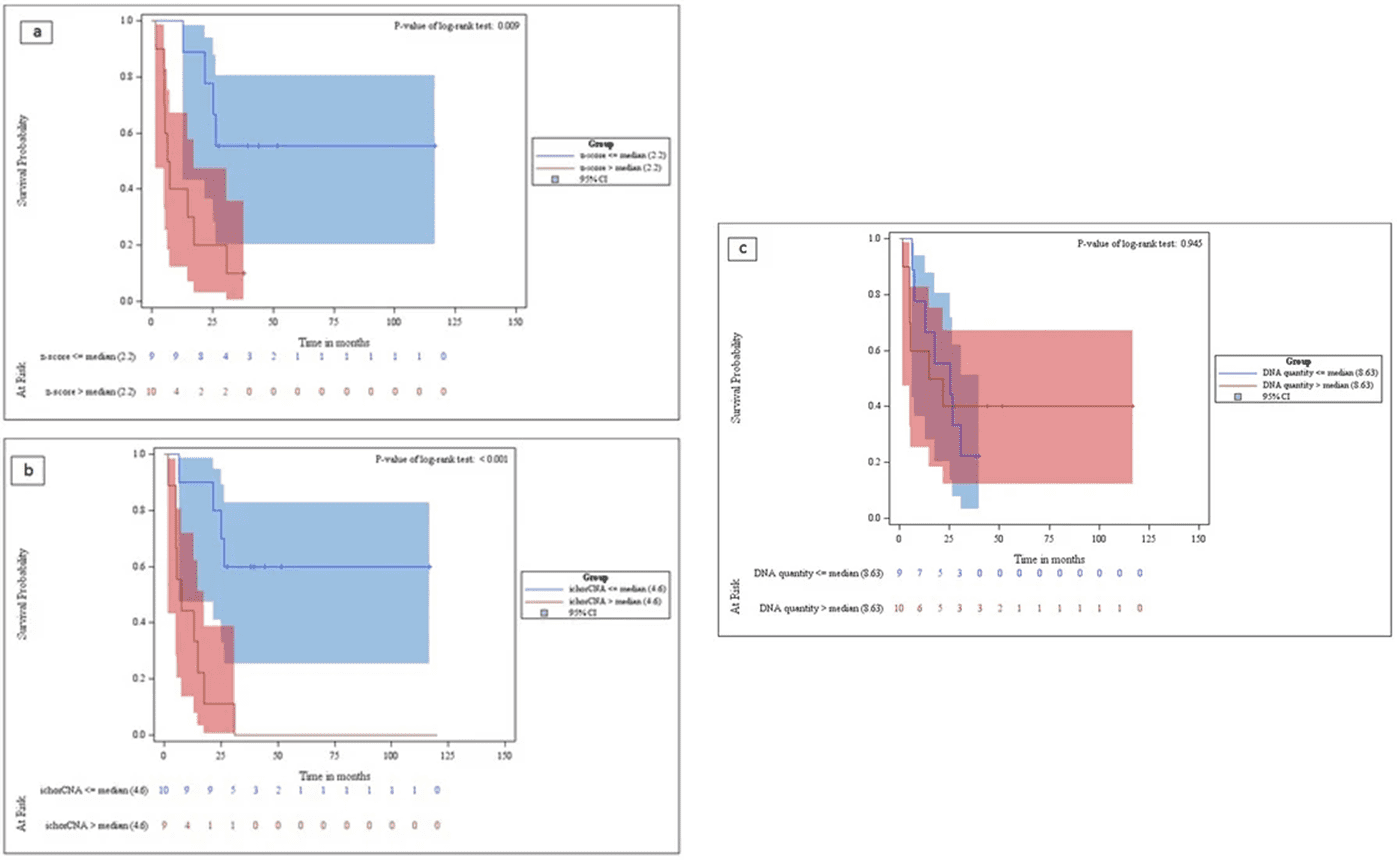 Fig 3. Kaplan–Meier analysis curves for the overall survival of metastatic breast cancer patients.
Fig 3. Kaplan–Meier analysis curves for the overall survival of metastatic breast cancer patients.
Conclusion
In summary, this study highlights the utility of shallow whole-genome sequencing (sWGS) for detecting clinically relevant somatic copy number alterations (SCNA), like FGFR1 amplification, in plasma of metastatic breast cancer patients. sWGS offers a valuable screening tool when fresh biopsies are unavailable, enabling reassessment of amplification status using archived samples. Additionally, both mFAST-SeqS and sWGS show promise for estimating tumor fraction and monitoring tumor dynamics in advanced cancer patients.
Reference:
- Bourrier C, Pierga J-Y, Xuereb L et al. Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival. Cancers. 2020; 12(6):1481.
Here are some publications that have been successfully published using our services or other related services:
Distinct functions of wild-type and R273H mutant Δ133p53α differentially regulate glioblastoma aggressiveness and therapy-induced senescence
Journal: Cell Death & Disease
Year: 2024
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Genome Analysis and Replication Studies of the African Green Monkey Simian Foamy Virus Serotype 3 Strain FV2014
Journal: Viruses
Year: 2020
See more articles published by our clients.


