Methods for microbial diversity studies
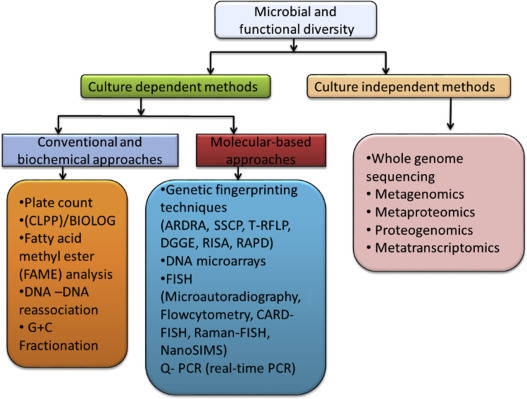
Methods for studying microbial diversity have been developed over the years and can be divided into traditional research methods and modern molecular biology methods. The traditional microbial community analysis method is based on microbial isolation and pure culture, and the community structure is understood through microscopic observation and physiological and biochemical characteristics study of pure microorganisms, and several non-culture methods are gradually developed in response to the limitations of culture methods to study the types and quantities of microorganisms. These methods are broadly classified as biochemistry, physiology, etc.
Traditional research methods
Traditional methods of microbial isolation and culture
The process of obtaining only one kind or one strain of microorganism from a mixed microbial population is called microbial isolation and purification. The principle is to select suitable for microbial growth of nutrients, temperature and other conditions for environmental microbial culture. The general process of this method is as follows: gradient dilution of the sample to the appropriate concentration, coating on the corresponding solid medium, culture at the appropriate temperature, single colony purification, 16S rRNA gene amplification sequencing identification, storage. However, due to the complex microbial community structure and high species diversity in the environment, the method of pure isolation and enrichment culture is not only time-consuming and laborious, but also has fatal methodological defects: (1) the unculturability of a large number of microorganisms in nature makes it impossible for humans to culture all microorganisms in nature; (2) The separation and enrichment culture method has strong selectivity, so that the microorganisms obtained by culture can not reflect the real situation of microbial communities in the natural state in terms of variety and function (Rossell-mora et al., 2001).
Phospholipid fatty acid (PLFA)
PLFA exists in cell membranes, and because different microorganisms have different types and quantities of PLFA, it can be used as an indicator to understand the changes in the composition and number of microbial species in the natural environment. However, PLFA model can not give an actual microbial species composition, only a general map of community structure.
Biolog
Biolog microplate identification system is a system developed by Biolog Corporation to determine the utilization of 95 carbon sources by microorganisms and to identify chemotrophic bacteria based on this system. However, because different microorganisms have different utilization ability of the same carbon source, the difference of microbial metabolic fingerprints to different single carbon source cannot be simply summarized as the difference of microbial community quantity and structure, so the accuracy of Biolog method is limited to some extent. But the Biolog method is popular because it is fast and easy.
Modern molecular biology methods
As far as microbes are concerned, the cultivable ones are less in numbers, while uncultured or non-cultivable have just started showing up with the modern tools and techniques. These techniques help to increase our knowledge about the diversity of genetic resources and understand the distribution of organisms, the fundamental role of diversity, the ongoing interactions between various useful or pathogenic microbial communities, and a wide array of possibilities.
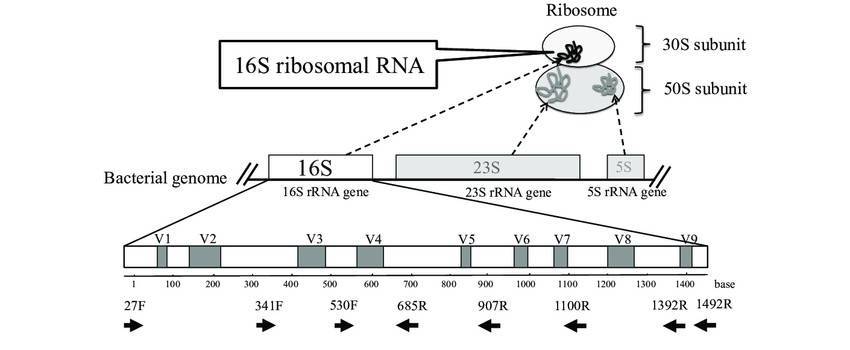
Modern molecular biology techniques mainly include denaturant gradient gel electrophoresis (DGGE)/temperature gradient gel electrophoresis (TGGE), restriction fragment length polymorphism (RFLP)/terminal restriction fragment length polymorphism (T-RFLP), Single Strand Conformation Polymorphism (SSCP), DNA microarrays, fluorescent in situ hybridization (FISH), metagenomic analysis, high-throughput technology etc.
Denaturant gradient gel electrophoresis/temperature gradient gel electrophoresis(DGGE/TGGE)
DGGE is used to analyze the diversity and relative richness of microorganisms by typing PCR products by electrophoresis. The principle of electrophoresis is used to separate DNA fragments by either a temperature or chemical gradient to denature the sample as it moves across an acrylamide gel. DNA segments which are same in length but have different base-pair sequences can be separated using these techniques. The separation is based on the difference in mobility of partially melted DNA molecules in acrylamide gels containing a linear gradient of DNA denaturants (urea and formamide). Hence, DNA sequences having a difference in only one base-pair can be separated by DGGE. TGGE employs the same principle as DGGE, but in this method, the gradient is temperature rather than chemical denaturants. DGGE was originally developed as a technique to study mutational sites in DNA sequences. In 1993, Muyzer et al. (1993) first applied DGGE to the study of microbial genetic diversity. PCR-DGGE a culture-independent approach that has been used to analyze microbial community structure across different fields, such as food microbiology, oral microbiology, soil microorganisms, environmental microbiology, and other areas (Sidira et al., 2014; de Paula et al., 2014; Jonathan et al., 2013; Zhong et al., 2014).
Restriction fragment length polymorphism/ terminal restriction fragment length polymorphism (RFLP/T-RFLP)
Another approach to study microbial diversity is restriction fragment length polymorphism (RFLP). PCR-RFLP is a combination of PCR technology, RFLP analysis and electrophoresis. First, the target DNA fragment to be detected is replicated and amplified, and then the amplified product is digested by DNA restriction endonuclease. Finally, it is analyzed by electrophoresis whether the target DNA fragment is cut or not. Another variant of this technique is terminal RFLP that addresses some of the limitations of RFLP by providing an alternate method for rapid analysis of microbial community diversity in different environments. Kanokratana P et al. (2004) used RFLP to study prokaryotic diversity in the Bor Khlueng hot springs, Thailand; Everroad R C et al. (2012) used T-RFLP analysis to study the effect of temperature on microbial diversity in Nakabusa hot springs, Japan, in order to better understand the biogeography and relationship between temperature and community structure within microbial mats. Based on the principal of RFLP, there are other techniques that were used sporadically. They are RISA and the automated version called ARISA. Here, the intergenic spacer region between the 16S and 23S ribosomal subunits is amplified by PCR and separated on a polyacrylamide gel under denaturing conditions. This is found to have differentiating potential for bacterial strains and closely related species (Fisher and Triplett, 1999).
Single strand conformation polymorphism (SSCP)
Single strand conformation polymorphism (SSCP) also relies on electrophoretic separation based on differences in DNA sequences and allows differentiation of DNA molecules having the same length but different nucleotide sequences. This technique was originally developed to detect known or novel polymorphisms or point mutations in DNA (Peters et al., 2000). In this method, single-stranded DNA separation on polyacrylamide gel was based on differences in mobility resulted from their folded secondary structure (Heteroduplex) (Lee et al., 1996). As formation of folded secondary structure or heteroduplex and hence mobility is dependent on the DNA. SSCP also suffers from the limitations as like DGGE, and hence the same DNA sequence can produce multiple bands on the gel. However, it does not require gradient gel and has been used for diversity studies (Stach et al., 2001).
Microarrays
Gene microarrays have been used to analyze the composition and diversity of microorganisms in samples for nearly 20 years, and have undergone several generations of improvement. The commonly used microarrays include PhyloChip (used to identify microorganisms and their phylogenetic relationships and analyze the diversity of microorganisms) and Functional GeoChip (used to study the diversity of functional genes and the activity of functional microorganisms). Microarray is a chip filled with probes (known short sequences for hybridization with sample DNA), which provide phylogenetic information or functional property information of organisms, or both. When the sequence in the sample (the fluorescence-labeled PCR product of the target fragment of DNA or RNA of pure bacteria and environmental samples or the fluorescence-labeled PCR product of random primers) hybridizes with the probe, the relative fluorescence ratio of the sequence matched with the probe can be calculated, so as to obtain the diversity and relative richness information of microorganisms. Gene microarray method is especially suitable for identifying the differences of representative microorganisms or microbial communities between different times, places and treatments. In addition, PhyloChip can also quantify the changes of related functional genes such as C, N, S and P cycles, degradation of organic pollutants and stress response. Aronson et al. (2013) used the third generation PhyloChip(16S rRNA gene microarray, which can provide about 60,000 different OTU information) to study the microorganisms related to methane cycle in pine forest soil under different experimental conditions.
Fluorescent in situ hybridization (FISH)
FISH is one of the most popular methods for DNA hybridization. Nucleic acid hybridization using specific probes is an important qualitative and quantitative tool in molecular bacterial ecology (Clegg et al., 2000). These hybridization techniques can be done on extracted DNA or RNA, or in situ. The sample is lysed to release all nucleic acids. Dot-blot hybridization with specific and universal oligonucleotide primers is used to quantify rRNA sequences of interest relative to total rRNA. The relative abundance may represent changes in the abundance in the population or changes in the activity and hence the amount of rRNA content (Theron and Cloete, 2000). Spatial distribution of bacterial communities in different environments such as biofilms can be determined using FISH (Schramm et al., 1996).
Metagenomic analysis
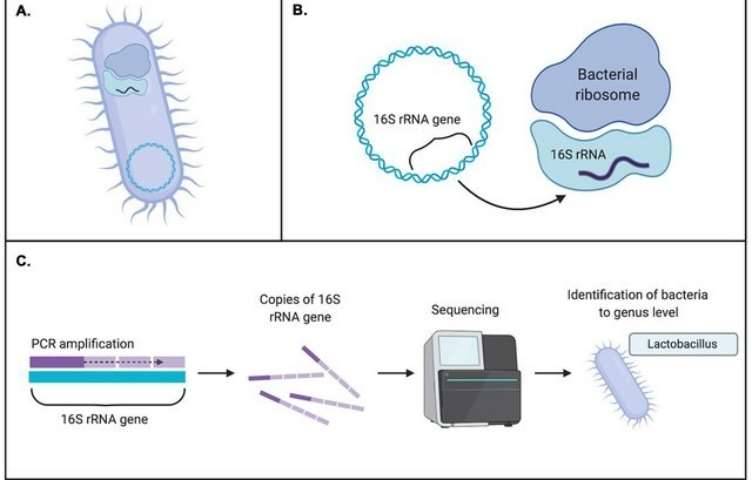
Metagenomics is defined as the functional and sequence-based analysis of the collective microbial genomes that are contained in an environmental sample (Zeyaullah et al., 2009). This contains all the genomes from coexisting microbes-called microbial communities. This is further subjected to the sequencing to know about the species composition in the sample. This provides a comprehensive view of the genetic diversity, species composition, evolution, and interactions with the environment of natural microbial communities (Simon and Daniel, 2011).
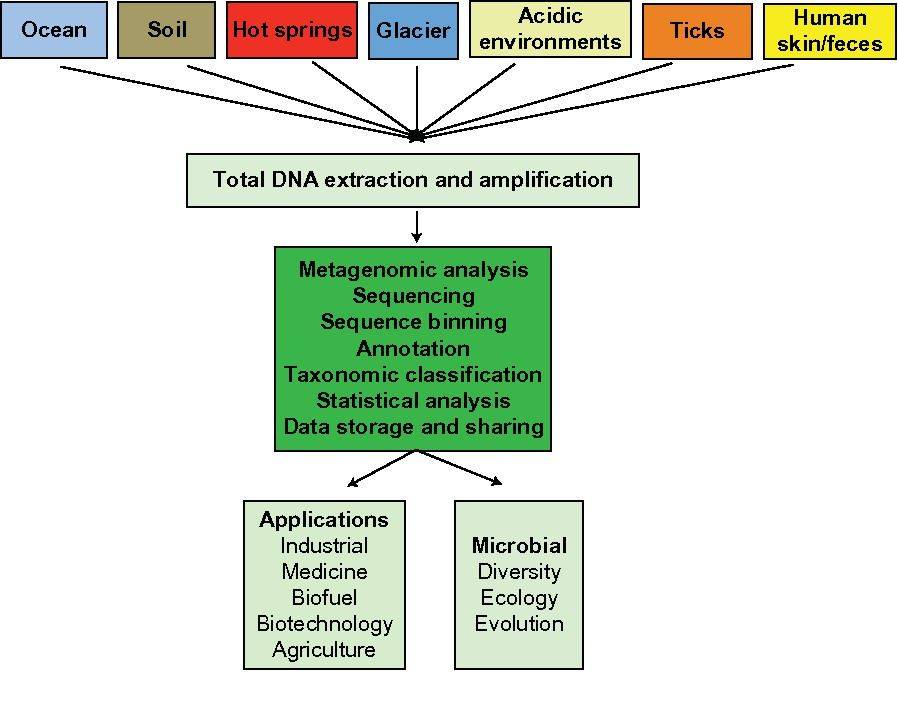 Overview of metagenomic analysis. (Girish Neelakanta et al,.2013)
Overview of metagenomic analysis. (Girish Neelakanta et al,.2013)
High-throughput technology
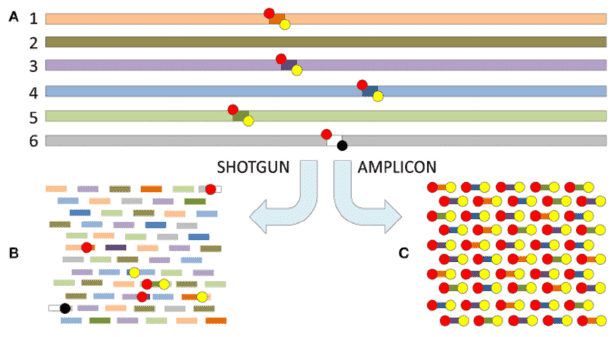
Sanger sequencing has been used for decades, and the sequencing quality is high, with a sequence length of 750—1000 bp. This method can't measure the mixed sequence, so it is necessary to establish a clone library first, that is, transfer the target sequence to the host cell for culture to form a single strain, and then sequence the target sequence in the single strain. It is time-consuming and expensive to analyze environmental samples with high microbial diversity.
On the basis of Sanger sequencing, high-throughput sequencing, also called next-generation sequencing (NGS), was developed. This method used PCR products of microbial target genes as samples to sequence, and tens of thousands to millions of sequences were obtained in one reaction, which greatly improved the breadth and depth of sequencing. At present, amplicon sequencing is commonly used methods for microbial diversity analysis in environmental samples, namely, 16S/18S/ITS sequencing. These high-throughput sequencing methods can identify the sequences of different samples by labeling the primers of each sample, and can simultaneously analyze multiple environmental samples and obtain a large number of sequences.
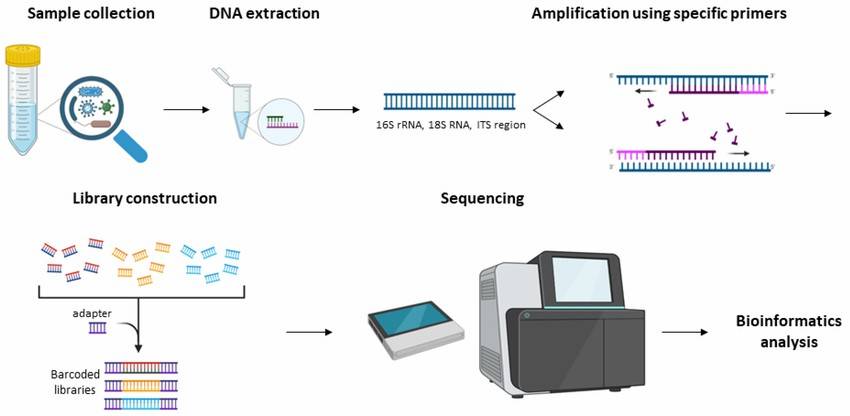 Demonstration of the workflow for the 16S rRNA and ITS amplicon sequencing.(Konstantina Athanasopoulou et al,. 2023)
Demonstration of the workflow for the 16S rRNA and ITS amplicon sequencing.(Konstantina Athanasopoulou et al,. 2023)