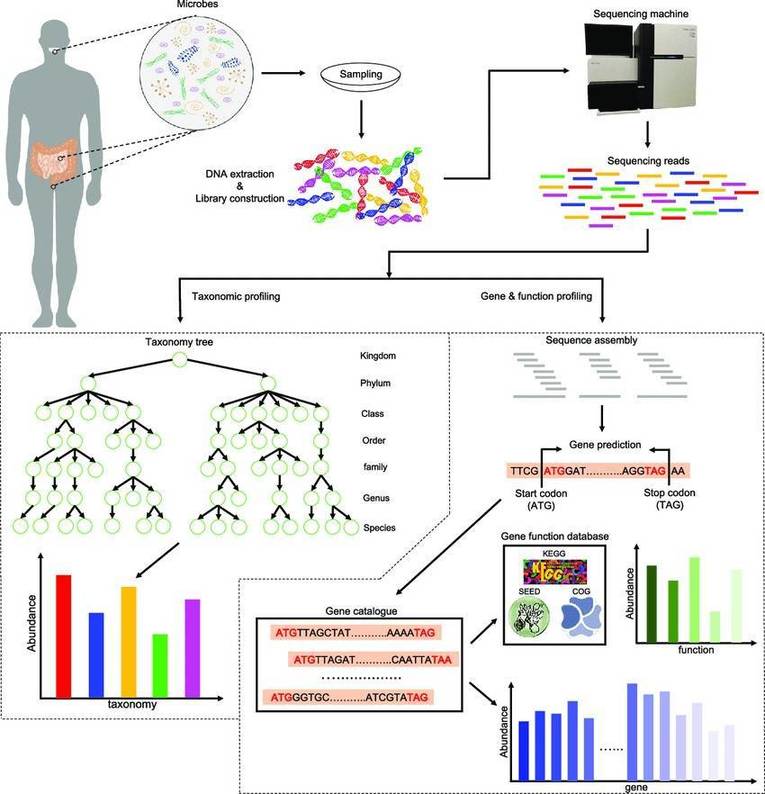
Backgrounds
In less industrialized nations, particularly Southeast Asia and sub-Saharan Africa, diarrheal diseases represent the third leading cause of mortality in children under five years of age. While recent improvements in sanitation and living standards have notably reduced the incidence and mortality rate of diarrheal diseases, childhood diarrhea resultant from viral infections remains a significant public health concern in developing countries. Viruses are the primary pathogens in such cases, accounting for 75% of total incidences. Array of viruses causing diarrhea includes Human Adenovirus (HAdV), Norovirus (NoV), Sapovirus (SaV), Human Astrovirus (HAstV), Rotavirus, Enterovirus, and Picornaviruses. However, knowledge about the prevalence of these viruses in children suffering from diarrhea remains considerably limited to date.
Methods
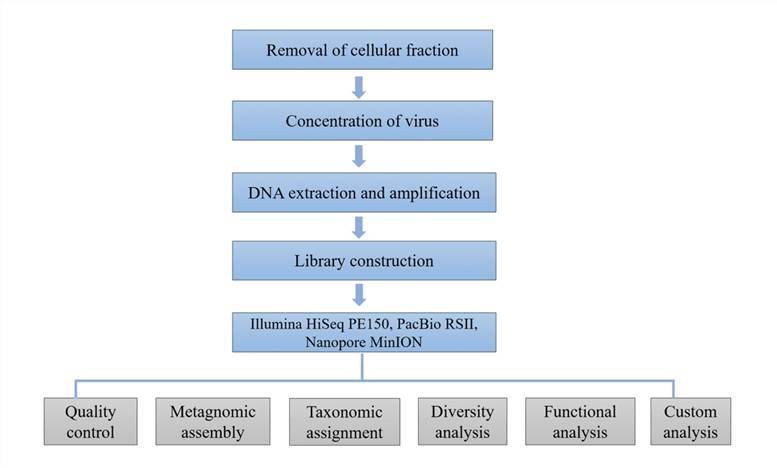
Sample preparation:
-
One hundred fresh stool samples
- Viral DNA and RNA extraction
Viral Metagenomics Sequencing:
-
Reverse transcription synthesizes
- Library construction
- Miseq Illumina platform
Results
1. Viral metagenomic overview
In this study, analysis of 100 fecal samples from children with diarrhea revealed that among the eukaryotic viral families, Caliciviridae predominated at 78.42%, followed by Adenoviridae (8.94%), Picornaviridae (8.36%), Reoviridae (2.81%), Astroviridae (1.18%), Reoviridae (0.20%), Anelloviridae (0.04%), and Picobirnaviridae (0.03%).
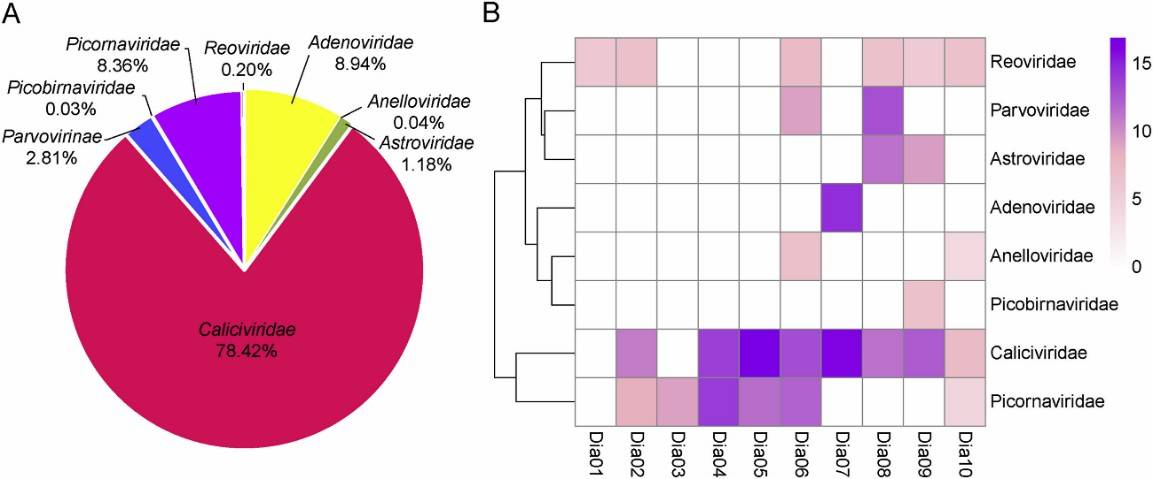 Fig. 1. Taxonomic analysis of fecal virome detected in diarrheal children on the family level. A The composition of fecal virome detected in diarrheal children. The percentage of virus sequences in different virus family was shown. B Heatmap representing the reads number of each viral family in exponential form.
Fig. 1. Taxonomic analysis of fecal virome detected in diarrheal children on the family level. A The composition of fecal virome detected in diarrheal children. The percentage of virus sequences in different virus family was shown. B Heatmap representing the reads number of each viral family in exponential form.
2. Phylogenetic analysis
In this study, nine viruses belonging to two genera of the Caliciviridae family were identified, of which eight were categorized as Norovirus (NoV), and one was part of the Sapovirus (SaV) genus. Seven viruses were under two genera of Picornaviridae, with three nearly full Enterovirus (EV) genomes and four almost complete sequences of Human parechoviruses. In addition, the presence of viruses belonging to the Mamastrovirus, Bocavirus, Mastadenovirus, and Rotavirus genera was also reported. This analysis expands the current knowledge of virodiversity in cases of paediatric diarrhoea.
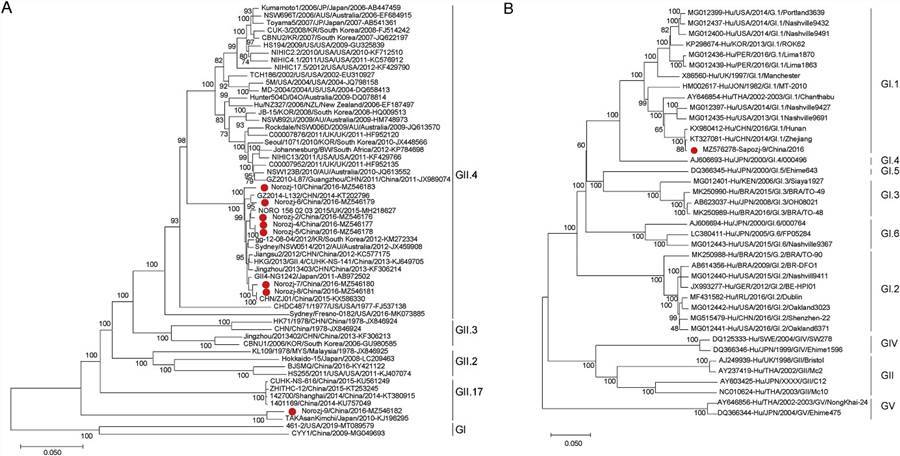 Fig. 2. The phylogenetic analysis based on the complete genome sequences of NoVs and SaVs.
Fig. 2. The phylogenetic analysis based on the complete genome sequences of NoVs and SaVs.
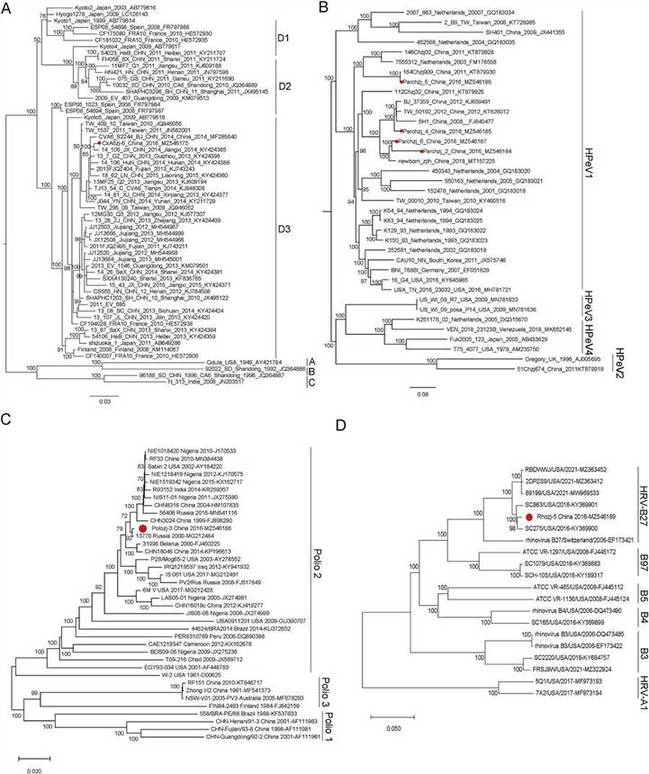 Fig. 3. The phylogenetic analysis of picornaviruses identified in this study.
Fig. 3. The phylogenetic analysis of picornaviruses identified in this study.
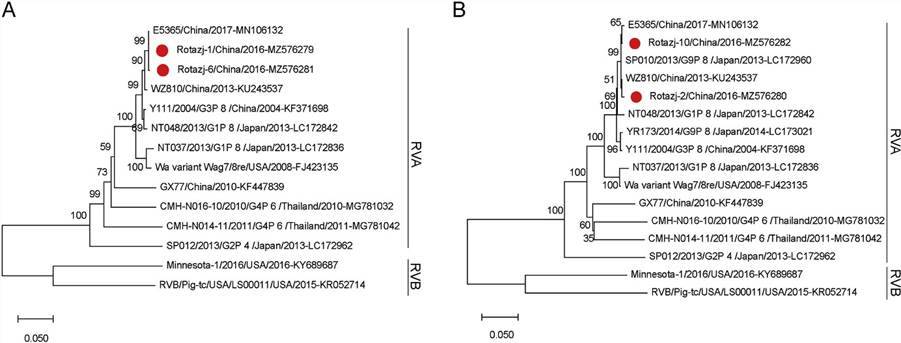 Fig. 4. The phylogenetic analysis of human rotaviruses identified in this study.
Fig. 4. The phylogenetic analysis of human rotaviruses identified in this study.
Conclusion
In this investigation, the researchers utilized viral metagenomic sequencing to analyze the viral communities present in fecal samples from children suffering from diarrhea. They observed that a significant proportion of DNA or RNA viruses associated with diarrhea belonged to six families: Adenoviridae, Astroviridae, Caliciviridae, Picornaviridae, Reoviridae, and Picobirnaviridae. Notably, Caliciviridae exhibited the highest prevalence at 78.42%, followed by Adenoviridae (8.94%) and Picornaviridae (8.36%). In addition to known pathogens implicated in human diarrheal diseases, the researchers identified unrelated viruses such as Circoviridae and Picobirnaviridae. This study advances our comprehension of the virome in pediatric diarrhea, furnishing valuable insights for the prevention and management of viral gastroenteritis.

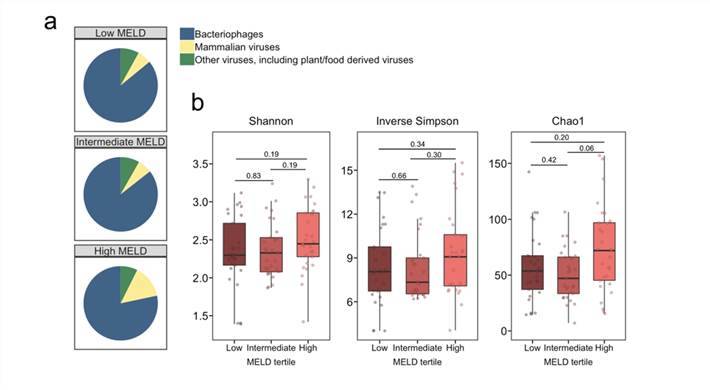
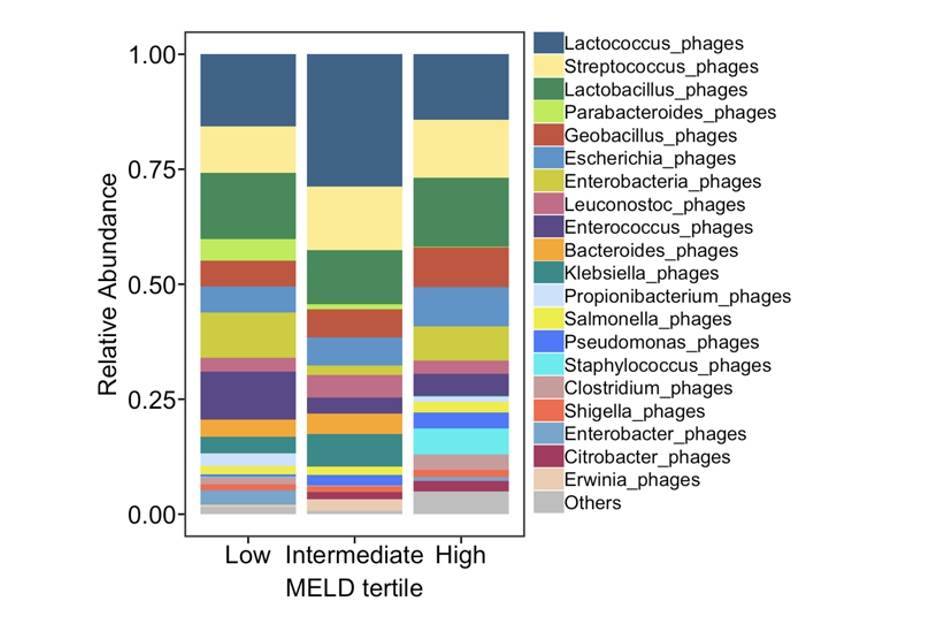 Intestinal virome in patients with alcoholic hepatitis. (Jiang et al., 2020)
Intestinal virome in patients with alcoholic hepatitis. (Jiang et al., 2020)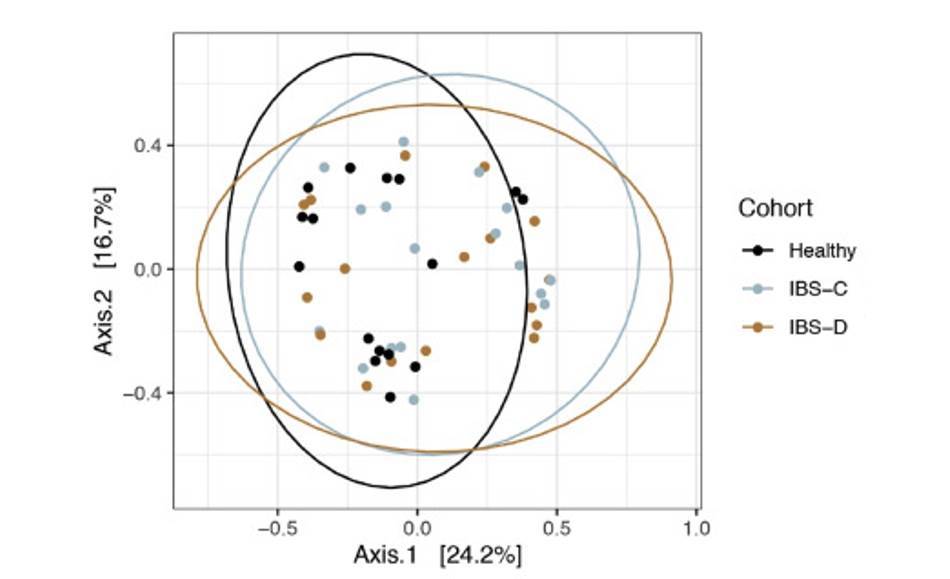 Multi-omics analyses show disease, diet, and transcriptome interactions with the virome. (Mihindukulasuriya et al., 2021)
Multi-omics analyses show disease, diet, and transcriptome interactions with the virome. (Mihindukulasuriya et al., 2021)