CD Genomics' Fungal ITS Sequencing provides precise species-level identification using optimized primers and advanced sequencing technologies. We deliver high-resolution data for detailed fungal community analysis, including diversity and phylogenetics. Our service combines cutting-edge technology with expert support to offer comprehensive insights into fungal ecosystems, aiding in research and ecological studies.
Our Advantages:
- High Resolution: Our sequencing approach delivers high-resolution data, allowing precise identification of fungal species and detailed characterization of fungal communities.
- Comprehensive Analysis: We provide a thorough analysis of fungal diversity, including species-level identification and community composition, which is crucial for ecological and clinical research.
- Advanced Technology: Utilizing cutting-edge sequencing platforms, such as Illumina, ensures high-quality data with minimal errors and high throughput.
- Expert Support: Our experienced team of biologists and bioinformaticians offers expert guidance and support throughout the entire sequencing process, from sample preparation to data interpretation.
What is an ITS Sequence
Internal Transcribed Spacer (ITS) sequences are critical components of ribosomal RNA (rRNA) genes in eukaryotes. These non-coding regions, specifically ITS1 and ITS2, are situated between the conserved coding regions of rRNA genes. In fungal genomes, ITS1 is located between the 18S and 5.8S rRNA genes, while ITS2 lies between the 5.8S and 28S rRNA genes. Notably, these regions exhibit considerable variability and are subject to fewer selective pressures compared to coding regions, making them particularly valuable for the study of fungal diversity and phylogeny. With lengths typically ranging from 500 to 750 base pairs, ITS sequences display significant sequence variability that mirrors evolutionary changes across fungal taxa. Consequently, ITS sequences serve as indispensable markers in fungal taxonomy, providing profound insights into the systematics and phylogenetic relationships among fungal species.
What is Fungal Sequencing
Fungal sequencing leverages advanced high-throughput sequencing technologies to analyze fungal DNA, including the ITS regions and the 28S rRNA gene. This process is integral for accurately identifying fungal species, understanding fungal community structures, and investigating their ecological roles within various environments.
Among the ITS regions, ITS1 and ITS2 are of particular importance. As non-coding regions, they experience less selective pressure and exhibit greater variability, providing the genetic diversity needed for detailed phylogenetic analyses. Recent studies suggest that ITS1 is often more effective than ITS2. ITS1 demonstrates higher efficiency, broader primer applicability, and a shorter amplification fragment with lower GC content compared to ITS2. Consequently, ITS1 may offer superior advantages for fungal sequencing, enhancing both the resolution and accuracy of fungal identification.
By utilizing NGS technologies, researchers can generate comprehensive profiles of fungal communities from environmental samples, clinical specimens, and other sources.
16S-ITS Association Study
Combining ITS and bacterial 16S rRNA sequencing, often referred to as the 16S-ITS association study, introduces a powerful framework for thoroughly analyzing microbial communities. 16S rRNA sequencing specializes in exploring bacterial populations. In contrast, ITS sequencing zeroes in on fungal communities. Merging these datasets, researchers achieve a panoramic view of the microbial ecosystem, revealing complex interactions between bacteria and fungi within a specific environment.
This integrative method significantly enhances our grasp of microbial diversity, ecosystem dynamics, and interspecies relationships. For instance, it can uncover how bacterial and fungal communities co-evolve, influencing each other's ecological roles. Such insights are critical for advancing our understanding of various contexts, be it environmental or health-related, providing a richer, more nuanced picture.
Applications
- Species Evolution
- Evolutionary Analysis
- Species Abundance
- Gene Discovery
- Environmental and Ecological Research
- Disease and Personalized Medicine
Service Specifications
Introduction to Our Fungal ITS Sequencing Service
At CD Genomics, our fungal ITS sequencing service utilizes advanced platforms like Illumina, PacBio, and Oxford Nanopore to deliver precise fungal identification and diversity analysis. Among these, the PacBio and Oxford Nanopore platforms are primarily used for Full-Length 16S/18S/ITS Sequencing. We ensure high-quality results through meticulous sample preparation and expert data analysis, providing detailed insights into fungal community structure and dynamics for applications in environmental and research settings.
Fungal ITS Sequencing Workflow
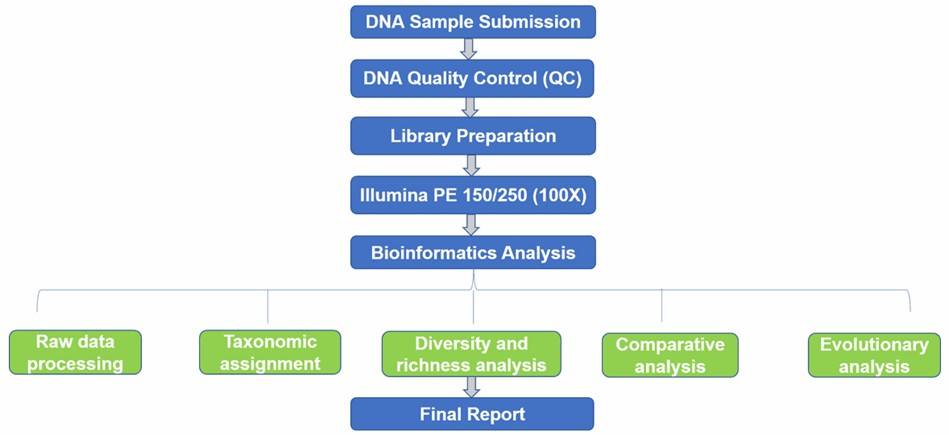
Technical Parameters
| Sequencing Platform |
Sequencing Strategy |
Data Volume |
| HiSeq, MiSeq |
PE300 |
Depending on the specific project requirements, not less than the contracted data volume |
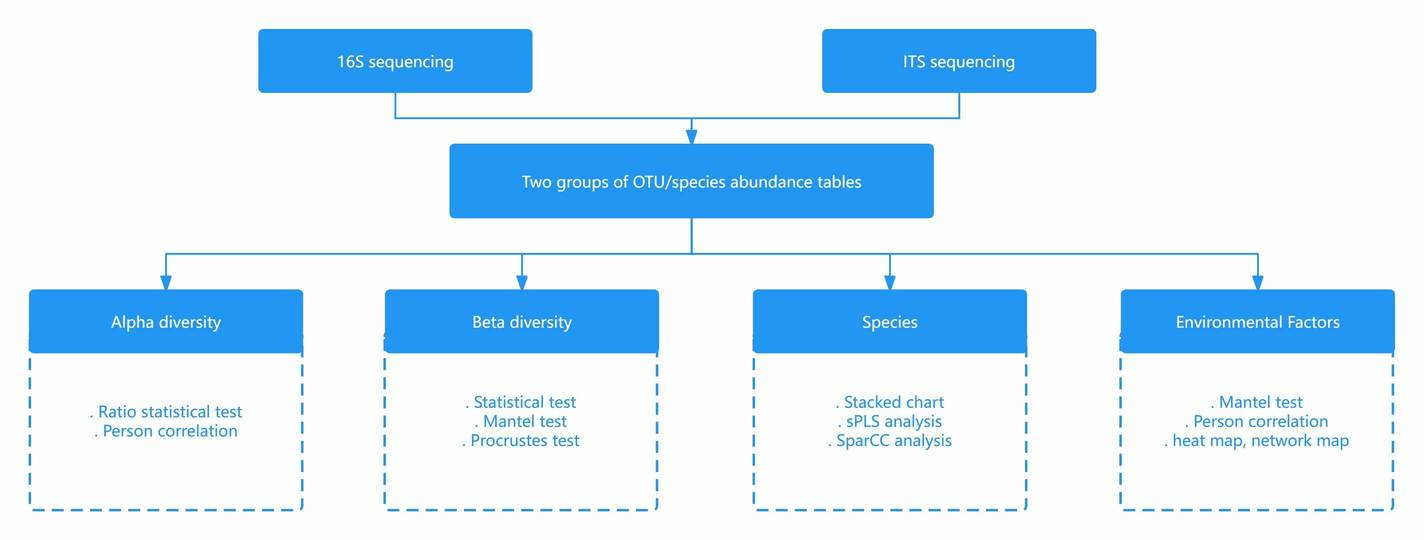
Bioinformatics Analysis
After DNA sequences are obtained, bioinformatics tools such as QIIME and mothur are commonly used for raw data processing, taxonomic assignment, diversity and richness analysis, comparative analysis, and evolutionary analysis. We are flexible to your needs.
| Pipeline |
Analysis Content |
| Raw data processing |
Filtering and trimming of poor-quality sequences, read alignment |
| Taxonomic assignment |
Operation Taxonomic unit (OTU) clustering and taxonomic assignment |
| Diversity and richness analysis |
Rank abundance, Venn, alpha diversity analysis, beta diversity analysis, rarefaction curves, heatmap, etc. |
| Comparative analysis |
Metastats, LEfSe, PCA analysis, PCoA analysis, etc. |
| Evolutionary analysis |
(Un)weighted UniFrac analysis, phylogenetic trees |
| Custom analysis |
CCA analysis, PICRUSt, network analysis, etc. |
Note: The above content includes only a portion of the bioinformatics analysis. For more information or to customize the analysis, please contact us directly.
Sample Requirement
| Sample Type |
Quantity |
Concentration |
OD260/OD280 |
| Tissue Sample |
>1.5 g |
- |
- |
| Genomic DNA |
≥200 ng |
≥6 ng/μl |
≥1.8 |
| PCR Product |
≥1 μg |
≥10 ng/μl |
- |
Sampling kits: We provide a complete range of microbial sampling kits for clients, including microbial collection products, DNA/RNA isolation kits, and accessories for storage and mailing.
Note: If you wish to obtain more accurate and detailed information regarding sample requirements, please feel free to contact us directly.
Deliverables
- Raw sequencing data (FASTQ)
- Trimmed and stitched sequences (FASTA)
- Quality-control dashboard
- Statistic data
- Your designated bioinformatics report.

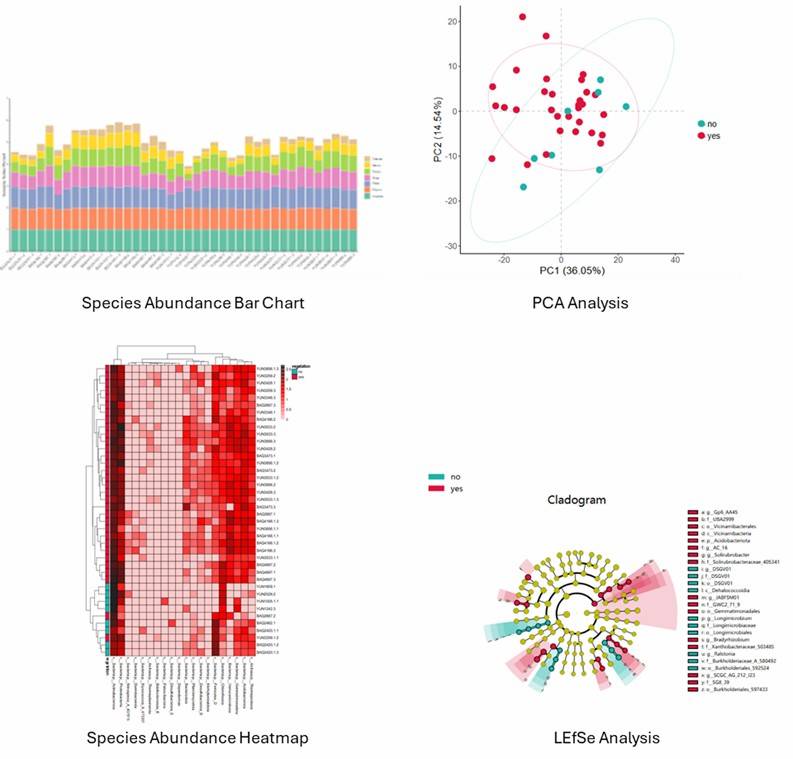

 Figure 1. Principal coordinates analysis (PCoA) of fungal composition based on weighted UniFrac in three groups.
Figure 1. Principal coordinates analysis (PCoA) of fungal composition based on weighted UniFrac in three groups.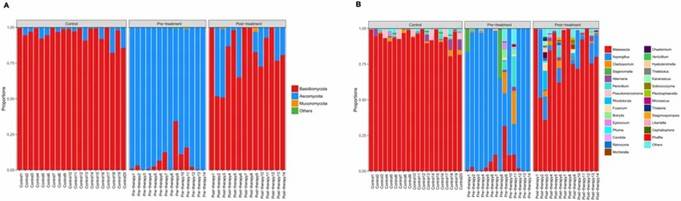 Figure 2. Distribution of predominant fungi in three groups at the phylum and genus levels.
Figure 2. Distribution of predominant fungi in three groups at the phylum and genus levels.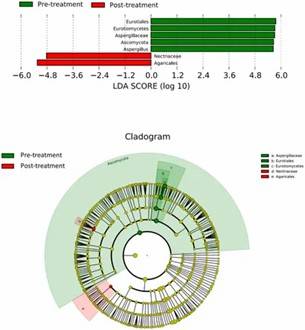 Figure 3. LEfSe analysis of fungal species. The bar graph shows the LDA scores calculated for characteristics at the OTU level.
Figure 3. LEfSe analysis of fungal species. The bar graph shows the LDA scores calculated for characteristics at the OTU level.