Traditional diagnostic techniques in microbiology, including isolation and cultivation, immunological assays, and microbial nucleic acid (DNA or RNA) detection, face challenges in meeting the dynamic and complex pathogen detection requirements in clinical settings due to limitations in timeliness, accuracy, and the restricted range of detectable pathogens. Metagenomic high-throughput sequencing (mNGS) stands out as a methodology that addresses these limitations. It involves high-throughput sequencing of all nucleic acids in a specimen, coupled with bioinformatic analyses, to identify pathogens comprehensively within the sample.
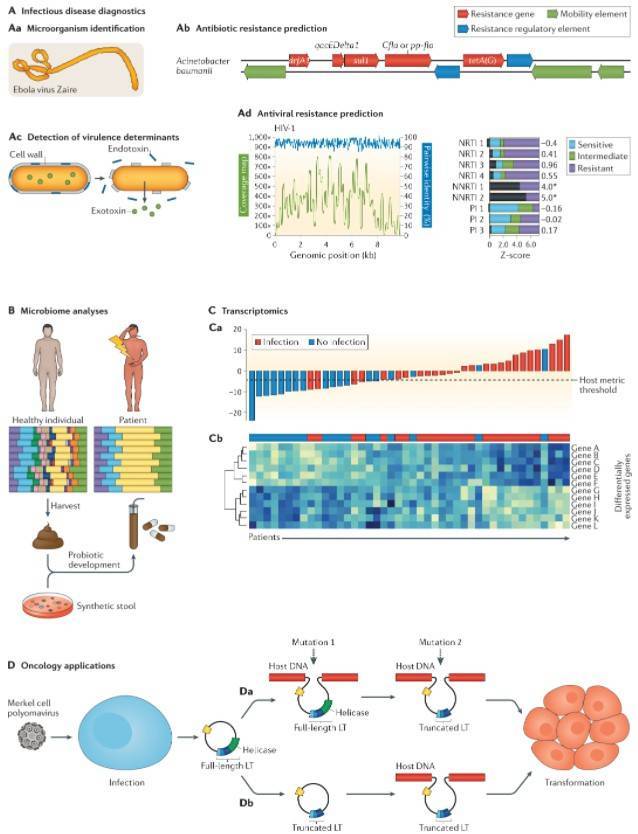 Clinical applications of metagenomic sequencing.
Clinical applications of metagenomic sequencing.
mNGS in Diagnosis of Infectious Diseases
Advancements in diagnostic techniques, particularly Metagenomic Next Generation Sequencing (mNGS), offer a powerful tool for identifying and analyzing pathogen genomes. This modern approach to diagnosing infectious disease has been increasingly adopted by independent medical laboratories worldwide. High-throughput sequencing-based pathogen detection is now frequently used in identifying pathogens across a wide array of infectious diseases; it has proven to be particularly potent in diagnosing pathogens linked with neurological infections, respiratory system infections, and bloodstream infections among others.
Metagenomic high-throughput sequencing found its initial application in the diagnosis of clinical neurological infections, leveraging an extensive spectrum of pathogens and sterile cerebrospinal fluid specimens. The sensitivity of mNGS in diagnosing pathogens in cerebrospinal fluid samples ranges from 73% to 92%, with a specificity of 96% to 99%. In the diagnosis of neurological infections, mNGS demonstrates a positive agreement rate of 80% when compared to conventional microbiological tests such as culture and immunological assays, while achieving an impressively high negative agreement rate of 98%.
Unlike sterile specimens such as cerebrospinal fluid, the respiratory and urogenital tracts harbor abundant normal microbial communities on their surfaces, posing challenges to mNGS detection. The sensitivity of mNGS in detecting pathogens in bronchoalveolar lavage fluid samples is reported to be 95.18%, with a specificity of 91.30%. However, due to methodological differences and variations in the definition of a "gold standard," the performance of mNGS in diagnosing respiratory tract infections varies significantly across studies, with reported sensitivities ranging from 47.9% to 100% and specificities from 41.7% to 100%.
Due to the systemic circulation of blood throughout the body, various infection sites can release a substantial amount of pathogenic nucleic acids into the bloodstream, facilitating the detection of infectious diseases. By examining microbial cell-free DNA (mcfDNA) in the blood, the application of mNGS extends beyond diagnosing bloodstream infections to include challenging scenarios where sampling is difficult or infections occur in deep-seated locations (e.g., infective endocarditis, invasive fungal infections). This broadens the scope of mNGS detection capabilities.
mNGS in Identification of Unknown Pathogens
mNGS displays an expansive capability for pathogen detection and the discovery of unknown pathogens, positioning it as an essential diagnostic tool to tackle emergent and sudden infectious diseases. Through the application of nanopore sequencing technology in metagenomics, scientists have accomplished real-time analyses of pathogens connected with emergent and sudden diseases, including the Ebola virus, Zika virus, and novel coronaviruses. This technology not only aids in monitoring epidemiological data but also enables the effective identification of viral mutation types, rendering significant support for epidemic prevention, control, and pathogen-based research.
mNGS in Antimicrobial Resistance Gene Detection
Microbial genomic data derived from mNGS not only serves the purpose of species identification but also facilitates the comprehensive analysis of antimicrobial resistance genes. However, the performance of mNGS in detecting antimicrobial resistance genes can be influenced by several factors, such as the types of pathogens, the kind of antibiotics, and the sequencing techniques employed. Experimental data has indicated that the accuracy rate of predicting antimicrobial resistance genes in patients with lower respiratory tract infections through mNGS falls between 78 - 87%. Studies have demonstrated that by integrating the CRISPR/Cas9 targeted enrichment strategy, the abundance of antimicrobial resistance genes can be amplified by 2500 times. Although the accuracy of predicting antimicrobial resistance genes with mNGS needs further assessment, the impediment of accurately detecting these genes may cease to be the major barrier for its application as the targeted enrichment techniques continue to evolve and the bioinformatics analysis processes are refined. Conversely, the interpretation of the results for antimicrobial resistance genes could pose as a significant challenge.
mNGS in Evaluating Host Transcriptomes
Emerging studies on mNGS integrate the identification of microbial species with host transcriptome data to create machine classification models, thereby significantly enhancing mNGS's capacity for diagnosing infectious diseases. By incorporating multifaceted information - such as the pathogen data, microbial diversity, and host transcriptomes detected by mNGS - into a diagnostic framework for lower respiratory tract infection, the negative predictive value reached a formidable 100%.
Diagnosing sepsis induced by immune dysregulation in the human body is another key application area where the integration of host-microbe mNGS analysis is proving invaluable. An integrated sepsis diagnostic model, developed using host and microbe information derived from mNGS testing of plasma specimens, was able to identify 99% of patients clinically diagnosed with sepsis.
Combining the high specificity of mNGS testing in detecting Mycobacterium tuberculosis with potent sensitivity provided by the host transcriptome classification model, the sensitivity and specificity in diagnosing tuberculous meningitis rose to 88.9% and 86.7% respectively. Hence, fully exploiting the pathogenic, microbial, and host transcriptome information obtained from a single mNGS test to enhance the accuracy of infectious disease diagnosis via mNGS embodies a critical direction for future development.
Conclusions and Future
The fast-paced development of mNGS technology in recent years, along with its widespread attention, has played a fundamental role in enhancing the diagnosis of infectious diseases, particularly in severe and complex cases. Yet, stumbling blocks to its clinical promotion include the lack of standardized testing methods, insufficient understanding and capability of personnel, and the high cost of equipment and reagents.
First and foremost, standardized testing is the bedrock for promoting the clinical application of mNGS. With the advent of a variety of new technologies and methods, there is an urgent need for additional methodological standardization studies to regulate the complete process of mNGS - from clinical indications to sample collection, testing, and result reporting.
Next, the imperative need in current clinical mNGS testing is the cultivation of compound talents who are well-versed in standard operating procedures, possess a certain level of bioinformatics analysis skills, and have background knowledge in clinical microbiology and clinical medicine. Enhancing the knowledge reserves and comprehensive capabilities of personnel is a priority.
Nevertheless, the potential for mNGS to identify all possible pathogens in a single test may prove more cost-effective than a series of traditional microbiology examinations. Therefore, there is a grave need for large-scale, prospective clinical research and economic data to assess the specific value of mNGS in improving the clinical management of patients with infectious diseases.
Fully integrating the pathogen information, transcriptome information, and drug-resistant gene information obtained from mNGS could help elevate the overall clinical management level for patients with infectious diseases - a paramount direction for mNGS development.
References
- Marais G, Hardie D, Brink A. A case for investment in clinical metagenomics in low-income and middle-income countries. Lancet Microbe, 2023, 4:e192-e199.
- Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet,2019,20: 341-355.
- Han D, Diao Z, Lai H, et al. Multilaboratory assessment of metagenomic next-generation sequencing for unbiased microbe detection. J Adv Res, 2022,38: 213-222.
- Miller S, Naccache SN, Samayoa E, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res, 2019,29: 831-842.
- Wilson MR, Sample HA, Zorn KC, et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. N Engl J Med, 2019,380:2327-2340.
- Diao Z, Lai H, Han D, et al. Validation of a Metagenomic Next-Generation Sequencing Assay for Lower Respiratory Pathogen Detection. Microbiol Spectr, 2022:e0381222.
- Diao Z, Han D, Zhang R, et al. Metagenomics next-generation sequencing tests take the stage in the diagnosis of lower respiratory tract infections. J Adv Res, 2022,38:201-212.
- Chien JY, Yu CJ, Hsueh PR. Utility of Metagenomic Next-Generation Sequencing for Etiological Diagnosis of Patients with Sepsis in Intensive Care Units. Microbiol Spectr, 2022,10: e0074622.
- Blauwkamp TA, Thair S, Rosen MJ, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol, 2019,4:663-674.
- Velmurugan G, Dinakaran V, Rajendhran J, et al. Blood Microbiota and Circulating Microbial Metabolites in Diabetes and Cardiovascular Disease. Trends Endocrinol Metab, 2020,31: 835-847.
- Lin B, Hui J, Mao H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors (Basel), 2021,11:214.
- Boolchandani M, D'Souza AW, Dantas G. Sequencing-based methods and resources to study antimicrobial resistance. Nat Rev Genet, 2019,20:356-370.
- Charalampous T, Kay GL, Richardson H, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol, 2019,37:783-792.
- Serpa PH, Deng X, Abdelghany M, et al. Metagenomic prediction of antimicrobial resistance in critically ill patients with lower respiratory tract infections. Genome Med, 2022,14:74.
- Langelier C, Kalantar KL, Moazed F, et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci U S A, 2018,115:e12353-e12362.
- Kalantar KL, Neyton L, Abdelghany M, et al. Integrated host-microbe plasma metagenomics for sepsis diagnosis in a prospective cohort of critically ill adults. Nat Microbiol, 2022,7:1805-1816.
- Ramachandran PS, Ramesh A, Creswell FV, et al. Integrating central nervous system metagenomics and host response for diagnosis of tuberculosis meningitis and its mimics. Nat Commun, 2022,13:1675.
- Han D, Li Z, Li R, et al. mNGS in clinical microbiology laboratories: on the road to maturity. Crit Rev Microbiol, 2019,45:668-685.