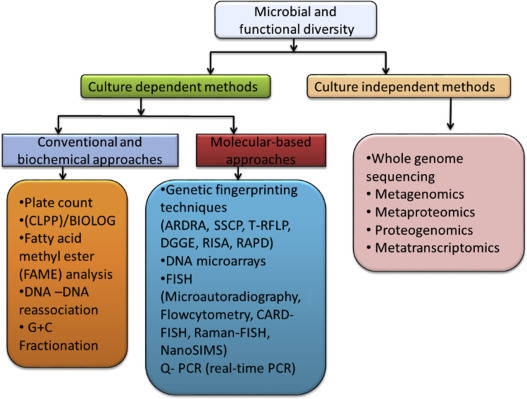
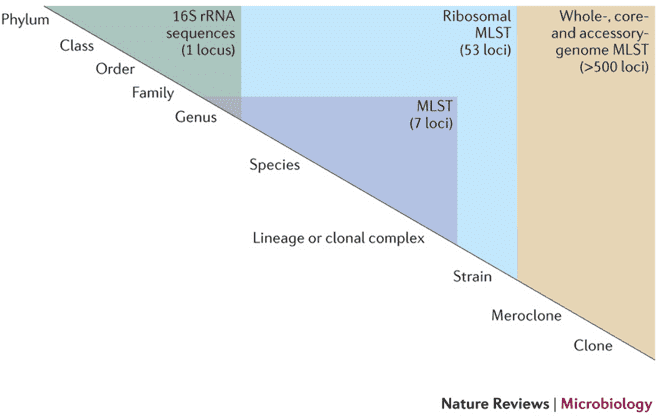
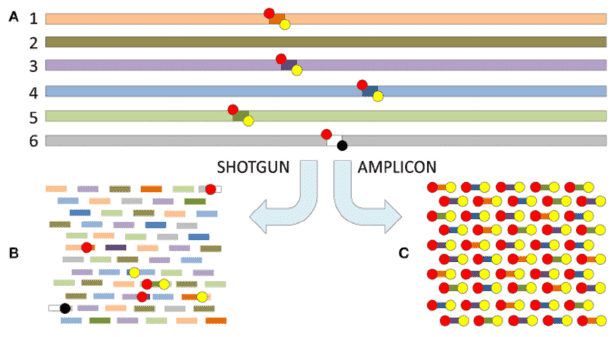
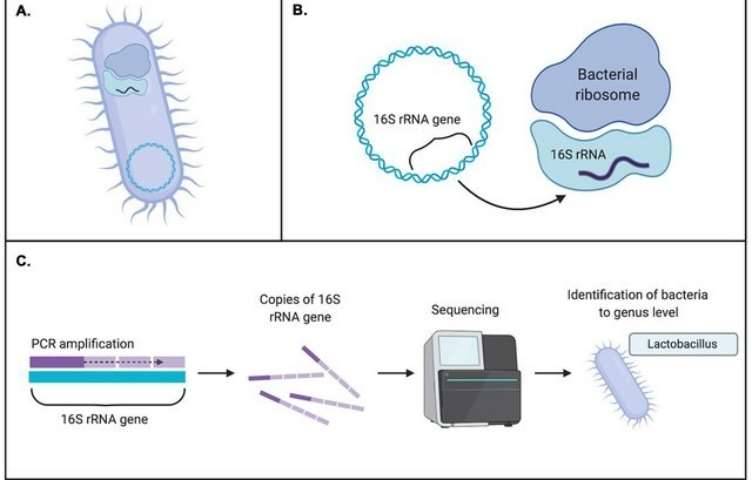
Why Microbial Diversity Analysis Matters
Microbial communities drive nutrient cycling, suppress diseases, and maintain ecosystem balance. Analyzing their diversity enables researchers to:
- Uncover microbial interactions and ecological roles
- Monitor shifts caused by environmental changes or interventions
- Support sustainable agriculture and improve soil health
- Deepen understanding of gut, plant, and marine microbiomes
How to Choose Your Microbial Diversity Sequencing Approach
Option 1: 16S/18S/ITS Amplicon Sequencing
Ideal for analyzing microbial community composition.
Technology Features:
- Illumina short-read sequencing targeting V3–V4 regions — cost-effective and high-throughput
- PacBio HiFi CCS long-read sequencing for full-length amplicons — delivers high-accuracy, strain-level resolution
Typical Applications:
Environmental microbiome surveys | Gut microbiome screening | Fungal community structure analysis
Provides simultaneous species identification and functional gene profiling.
Technology Features:
- Illumina short-read sequencing — high throughput for functional gene annotation
- Long-read sequencing — enables microbial genome reconstruction
Typical Applications: Antibiotic resistance gene studies | Microbial metabolic pathway analysis | Discovery of novel microbial genomes
Note: This page primarily covers 16S/18S/ITS amplicon sequencing for microbial community profiling. For combined species and functional gene insights, explore our metagenomic sequencing services.
Our 16S/18S/ITS Sequencing Services at a Glance
We provide targeted and full-length amplicon sequencing solutions designed to meet diverse microbiome research needs:

High-throughput analysis of bacterial diversity focusing on V3–V4 or other key variable regions.
Get Service Info
Accurate profiling of archaeal communities from extreme or specialized habitats.
Get Service Info
Identification of actinomycete subgroups critical for soil health, antibiotic production, and bioremediation.
Get Service Info
High-resolution profiling of fungal communities by targeting ITS1 or ITS2 regions.
Get Service Info
Comprehensive detection of eukaryotes, including protists, fungi, and microfauna.
Get Service Info
Long-read PacBio sequencing for enhanced taxonomic resolution and detailed phylogenetic insights.
Get Service Info
High-precision microbial fingerprinting ideal for complex or low-biomass samples.
Get Service Info
Research Applications & Recommended Sequencing Services
Tailored sequencing solutions for diverse microbiome research
| Research Application |
Recommended Services |
| Environmental Microbiome Profiling
|
- Bacterial 16S rRNA Sequencing
- Archaeal 16S rRNA Sequencing
- Fungal ITS Sequencing
- Eukaryotic 18S rRNA Sequencing |
| Host-Associated Microbiome Studies |
- Full-Length 16S/18S/ITS Sequencing
- 2bRAD-M Analysis |
| Low-Abundance or Complex Communities |
- 2bRAD-M Analysis
- Full-Length 16S Sequencing |
| Functional and Spatial Analysis |
- 2bRAD-M Analysis |
| Industrial or Fermentation Microbiome
|
- Actinomycetes Diversity Analysis
- Combined 16S and ITS Amplicon Sequencing |
Not sure which approach fits your study? [Contact our experts] for personalized guidance.
End-to-End Workflow: Tailored Solutions from Sample to Insight
Step 1: Project Consultation
Personalized 1-on-1 experimental design tailored to your research goals
Step 2: Sample Quality Control
DNA integrity assessment
Concentration normalization to ensure optimal sequencing input
Step 3: Sequencing & Data Analysis
Dual-platform sequencing using Illumina and PacBio
Quality benchmarks: Illumina Q30 ≥ 90%, PacBio CCS accuracy ≥ 99%
Step 4: Report Delivery
Comprehensive package including raw sequencing data and detailed analysis report
Request Quote
Technology Platforms We Use

Illumina NovaSeq / MiSeq / PE250
Short-read, high-throughput sequencing for diverse amplicon targets

PacBio SMRT (HiFi CCS)
Long-read, high-fidelity sequencing enabling full-length amplicon analysis

qPCR / Sanger
Validation and spatial detection methods to complement sequencing data
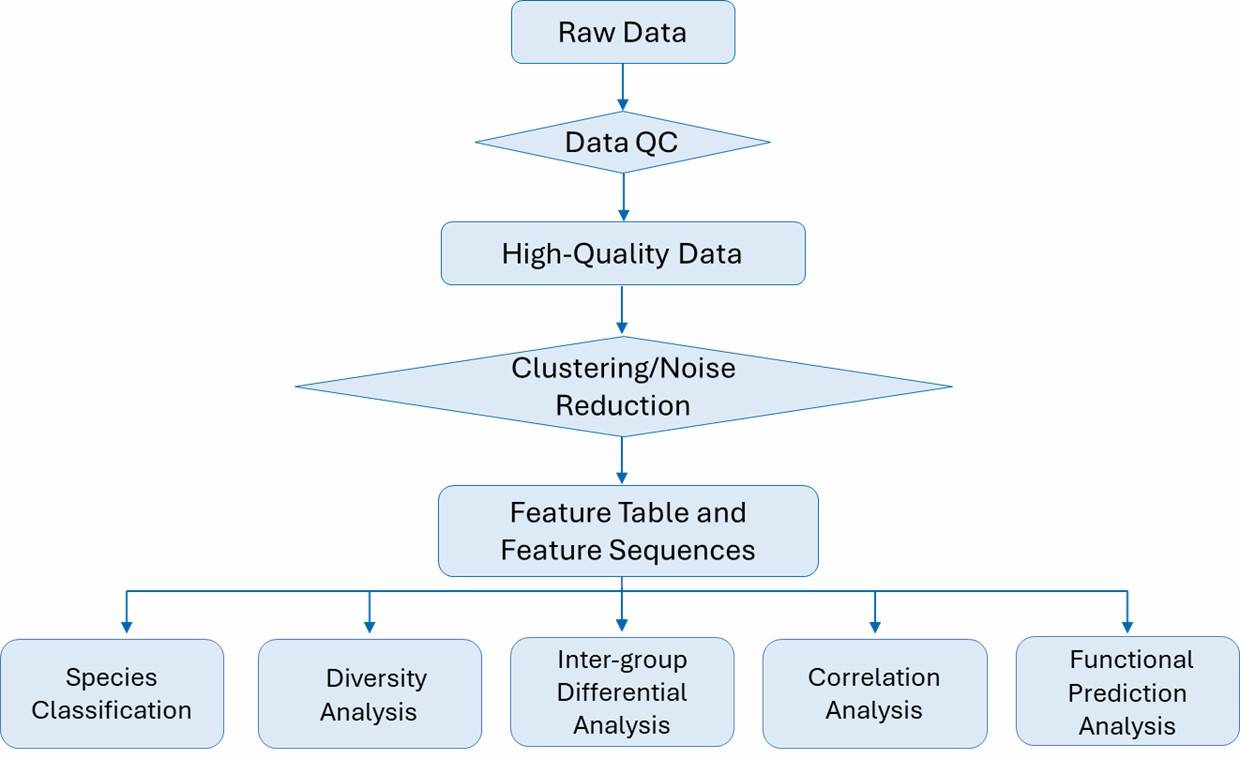
Comprehensive Bioinformatics Analysis
Our advanced bioinformatics pipeline delivers deep insights into microbial communities, supporting detailed taxonomic, functional, and ecological profiling. With stringent quality control and cutting-edge algorithms, we provide accurate, publication-ready results tailored to your research objectives.
Core Bioinformatics Analyses
| Analysis Type |
Key Methods |
Research Focus |
| Species Annotation |
DADA2 (ASV) / UPARSE (OTU) |
Identification of bacteria, fungi, and archaea |
| Alpha Diversity |
Shannon/Chao1 indices, rarefaction curves |
Species richness and evenness within samples |
| Beta Diversity |
PCoA / NMDS + PERMANOVA |
Differences in microbial community structure between groups |
| Differential Abundance |
LEfSe / ANCOM |
Detecting significantly different taxa across groups |
| Functional Prediction |
PICRUSt2 / FAPROTAX |
Inferring potential metabolic functions of microbes |
| Network Analysis |
SparCC / MENA |
Exploring co-occurrence and exclusion relationships among microbes |
Advanced Bioinformatics Options
- Antibiotic Resistance Gene (ARG) Analysis: Gene annotation and visualization using the CARD database
- Environmental Correlations: CCA/RDA analyses linking microbial profiles to physicochemical factors
- Time-Series Analysis: Tracking microbial community dynamics over multiple time points
Sample Requirement
| Service |
Sample Type |
Recommended Quantity |
Minimum Quantity |
Concentration |
| 16S/18S/ITS Sequencing |
Genomic DNA |
≥100 ng |
10 ng |
≥1 ng/μl |
| Full-Length 16S/18S/ITS Amplicon Sequencing |
Genomic DNA |
≥ 500ng |
|
10 ng/µL |
| Tissue |
1-3g |
1 g |
|
| Thallus |
5 g |
3 g |
|
| Interstitial Fluid |
3-5 mL |
1 mL |
|
| Environmental Samples |
3-5g |
1 g |
|
| Water filter membrane |
3 |
1 |
|
Note: If you wish to obtain more accurate and detailed information regarding sample requirements, please feel free to contact us directly.
Demo
Partial results of our microbial diversity analysis – 16S/18S/ITS sequencing service are shown below:
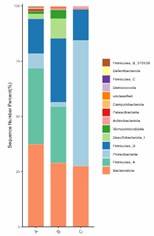
Microbial Distribution Bar Chart
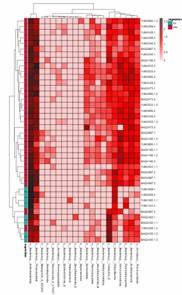
Classification Heatmap
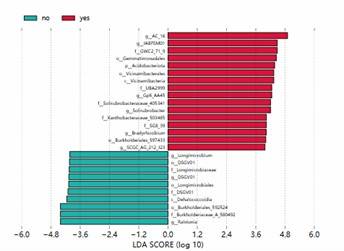
LEfSe Analysis LDA Bar Chart
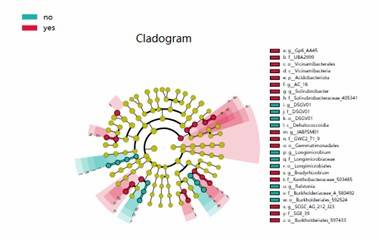
LEfSe Analysis Cladogram
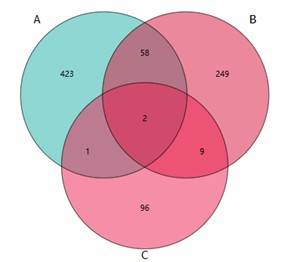
Venn Diagram
Beta Diversity Index Heatmap
Customer Case
Microbiota of the Digestive Gland of Red Abalone (Haliotis rufescens) Is Affected by Withering Syndrome
Journal: Microorganisms
Impact factor: 4.26
Published: 2020
Find out more
Backgrounds
The study centers on Withering Syndrome (WS), an infectious disease that heavily impacts abalone aquaculture. This disease is caused by the intracellular bacterium Candidatus Xenohaliotis californiensis, which infects the digestive glands, disrupting their function and causing progressive wilting in abalones. While the direct effects of WS are understood, its impact on the microbiota of the digestive glands in abalones is not well-studied. The main goal of this research is to determine if there are differences in the digestive gland-associated microbiota between healthy red abalones and those affected by WS.
Materials & Methods
Sample preparation:
-
Red abalone
- Converted land samples
- DNA extraction
Data Analysis:
-
Statistical analysis
- Beta diversity measures
- Community composition testing
- Differential abundance analysis
Results
The study found notable differences in microbiota composition between healthy and WS-affected abalones. Healthy abalones exhibited a diverse and balanced bacterial community, whereas WS-affected abalones had a microbiota dominated by specific bacteria associated with the disease. One of the most significant findings was that a certain bacterium was dominant in healthy abalones, while the primary pathogen of WS was more prevalent in the affected group. Interestingly, the pathogen was also present in some healthy specimens, indicating that the balance between different bacterial populations might be crucial in determining the disease status.
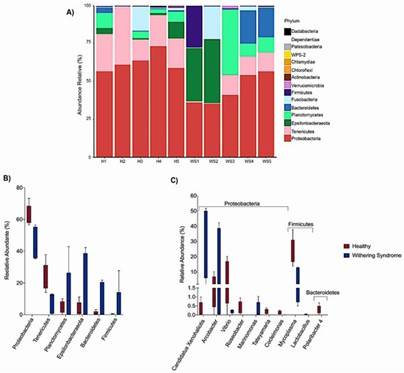 Figure 1. (A) Relative abundance of microbiota at the phylum level in the digestive glands of healthy (H1-H5) and withering syndrome-affected red abalone (WS1-WS5). (B) Comparison of relative abundance at the phylum level between healthy (red boxes) and affected (blue boxes) abalone. (C) Comparison at the genus level.
Figure 1. (A) Relative abundance of microbiota at the phylum level in the digestive glands of healthy (H1-H5) and withering syndrome-affected red abalone (WS1-WS5). (B) Comparison of relative abundance at the phylum level between healthy (red boxes) and affected (blue boxes) abalone. (C) Comparison at the genus level.
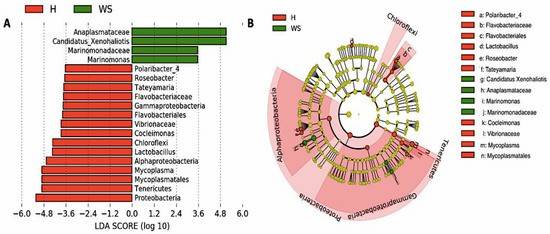 Figure 2. Differences in digestive gland microbiota of healthy red abalones (H) compared with red abalones with withering syndrome disease (WS).
Figure 2. Differences in digestive gland microbiota of healthy red abalones (H) compared with red abalones with withering syndrome disease (WS).
Conclusions
The findings suggest that WS significantly disrupts the microbiota composition in the digestive glands of red abalones. Specific bacterial communities are associated with either health or disease, highlighting the potential ecological role of microbiota in WS pathogenesis. Further research is needed to deepen our understanding of the dynamics of digestive gland microbiota and its influence on the progression of Withering Syndrome in abalones.
Published Research Supported by Our 16S/18S/ITS Sequencing Services
Explore representative publications by our clients, showcasing the real-world applications of 16S/18S/ITS sequencing in fields like agriculture, environment, aquaculture, and food safety.
Elucidating the effects of organic vs. conventional cropping practice and rhizobia inoculation on rhizosphere microbial diversity and yield of peanut
Paudel, Dev, et al. | Environmental Microbiome | 2023 | https://doi.org/10.1186/s40793-023-00517-6
Multi-species biofilms of environmental microbiota isolated from fruit packing facilities promoted tolerance of Listeria monocytogenes to benzalkonium chloride
Rolon, M. Laura, et al. Biofilm 2024 https://doi.org/10.1016/j.bioflm.2024.100177
Evaluating the impact of the biocontrol agent Trichoderma harzianum ITEM 3636 on indigenous microbial communities from field soils
Ganuza, M., et al. Journal of Applied Microbiology 2019 https://doi.org/10.1111/jam.14147
Exploring actinobacteria associated with rhizosphere and endosphere of the native alpine medicinal plant Leontopodium nivale subspecies alpinum
Oberhofer, Martina, et al. Frontiers in Microbiology 2019 https://doi.org/10.3389/fmicb.2019.02531
FAQ
- Q: What types of microbes do 16S, 18S, and ITS sequencing target?
- Q: Should I choose V3–V4 or full-length 16S?
- Q: Can 18S replace ITS for fungal sequencing?
- Q: How many samples do I need for reliable results?
- Q: What's the difference between OTUs and ASVs?
- Q: What insights do α and β diversity analyses provide?
- Q: What does the LDA score in LEfSe results indicate?
- Q: How do I identify microbial biomarkers from the data?
- Q: How should I process samples from extreme environments (e.g., high salt, high temperature)?
- Q: How can I reduce host DNA interference in plant endophyte studies?
- Q: How should I design a time-series microbiome study?