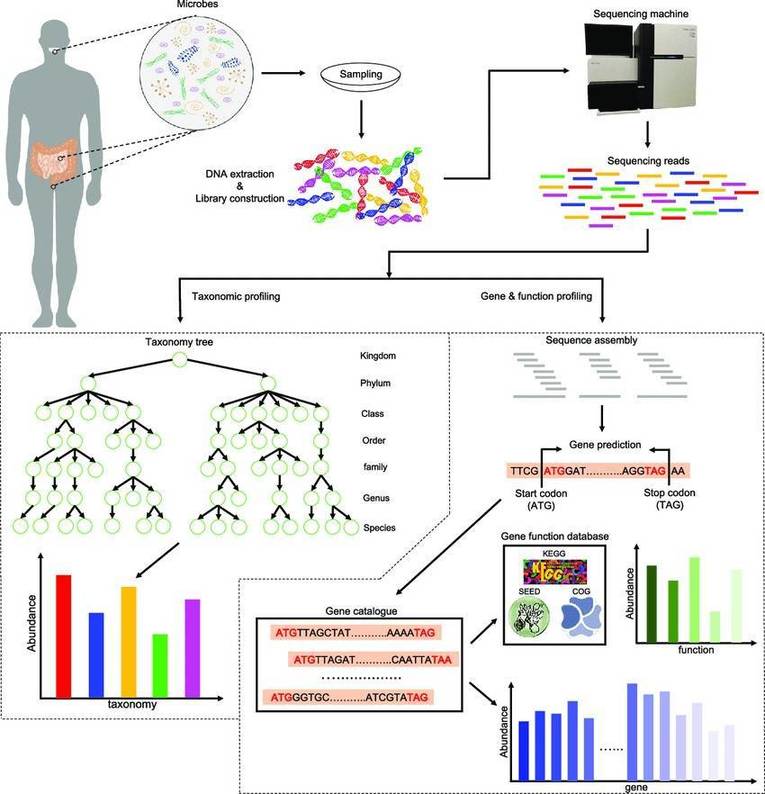
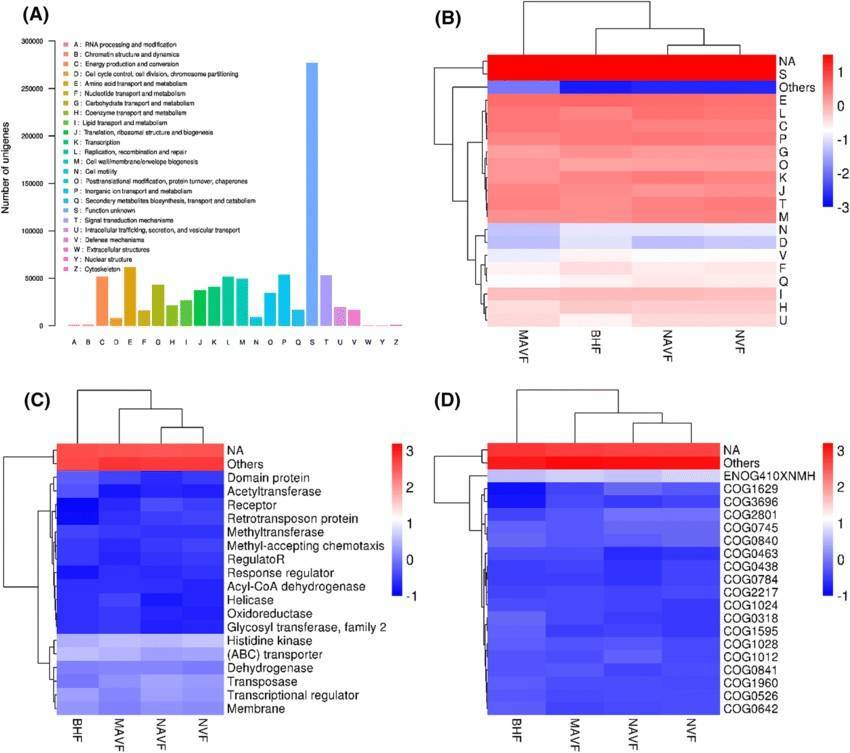
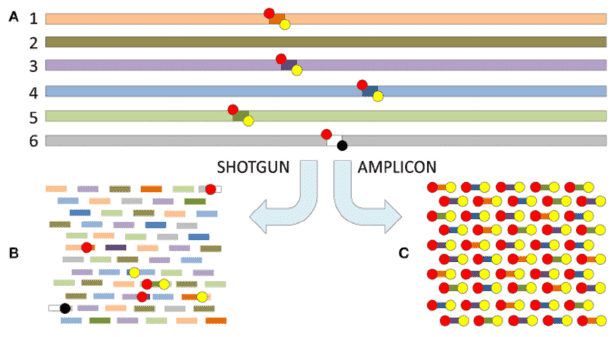
Our shotgun metagenomic sequencing platform stands as an invaluable resource, enabling customers to acquire intricate microbial genetic data from environmental samples. We are dedicated to delivering high-quality data, complemented by tailored bioinformatics analyses, to fulfill the specific requirements of our clients.
Our Advantages:
- 10+ years of commercial experience in life science research industry
- State-of-the-art sequencing platforms including both next-generation and long-read sequencing platforms
- Allow for the detection of low abundance members of microbial communities
- Flexible sequencing strategies and comprehensive data analysis
- Cost-effective and time-efficient services
- Satisfactory customer service throughout the whole service lifecycle
What is Shotgun Metagenomic Sequencing
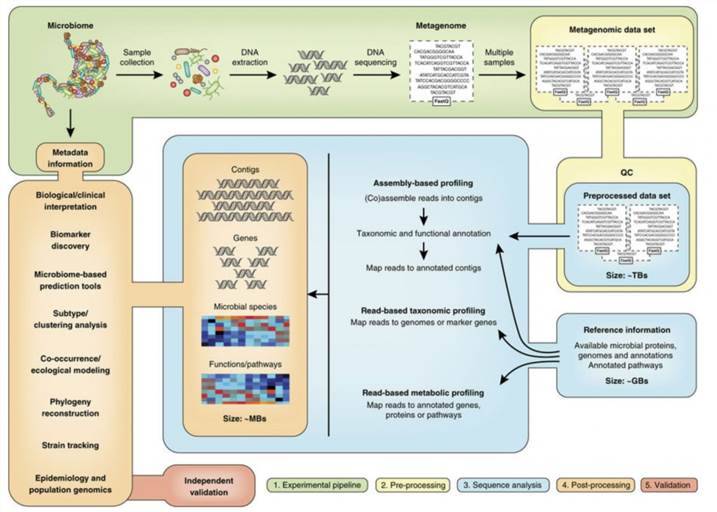
Shotgun metagenomic sequencing represents a sophisticated, untargeted methodology that capitalizes on next-generation sequencing (NGS) technologies to elucidate the complete genetic landscape of microbial communities within diverse environments. In contrast to traditional sequencing techniques, which predominantly target specific genes or organisms, shotgun metagenomics facilitates a comprehensive examination by capturing and analyzing the entire genomic complement present in a sample. This methodological approach is particularly advantageous for investigating intricate ecosystems such as terrestrial soils, aquatic habitats, and the human microbiome, where a multitude of microorganisms coexist and interact.
Moreover, shotgun metagenomic sequencing can also integrate third-generation sequencing platforms, which provide extended read lengths and heightened resolution. This technological refinement significantly enhances our capacity to resolve complex genomic architectures and identify previously uncharacterized microbial taxa. Consequently, it broadens our understanding of microbial diversity and functionality within their respective ecosystems, offering profound insights into microbial ecology and evolution.
Difference Between Shotgun Metagenomics and NGS
In the realm of microbial research, two cornerstone methodologies, amplicon-based NGS and shotgun metagenomic sequencing, are engineered for distinct investigative frameworks. Amplicon sequencing zeros in on specific genetic loci, exemplified by the 16S rRNA gene in bacteria and archaea, allowing for a precise elucidation of the taxonomic architecture within microbial communities. This targeted modus operandi is lauded for its cost-efficiency and the robustness of established protocols for library preparation and data analysis.
In contrast, shotgun metagenomic sequencing presents a broader perspective by undertaking an untargeted exploration of the entire genomic landscape within a sample. This approach reveals a wider array of microorganisms, including those that are unculturable and lack well-characterized genetic markers. Furthermore, it enables a comprehensive analysis of functional genes, providing deep insights into the ecological roles and interactions that characterize complex microbial ecosystems.
The selection of either amplicon-based or shotgun metagenomic sequencing hinges significantly on the research objectives at hand. When the aim is to delineate the structural delineations of specific microbial taxa, amplicon sequencing suffices admirably. Yet, in quests to decode intricate microbial interplay, unearth novel species, or elucidate the comprehensive functional dynamics within microbial consortia, shotgun metagenomic sequencing ascends as the more illuminating and informative strategy.
How is Shotgun Metagenomics Used to Study Infectious Diseases
The integration of shotgun metagenomic sequencing into infectious disease research has revolutionized pathogen identification, transmission tracking, and the monitoring of antibiotic resistance. Unlike traditional diagnostic methods, this cutting-edge technology uncovers previously undetected pathogens, offering a more comprehensive view of infectious agents and their behaviors.
In recent investigations, metagenomic next-generation sequencing (mNGS) has been pivotal in diagnosing community-acquired pneumonia (CAP). The superior detection capabilities of mNGS are particularly valuable in cases of mixed infections, where the presence of multiple pathogens complicates both diagnosis and therapeutic strategies.
Moreover, shotgun metagenomic sequencing has played a crucial role in the surveillance of infectious disease outbreaks. Its application in monitoring the mpox virus (MPXV), for instance, has yielded vital data on transmission routes and genomic mutations, thereby informing and optimizing public health responses.
Applications of Shotgun Metagenomic Sequencing
The applications of antibiotic resistance genes screening include, but are not limited to, the following areas:
- Medical Field: Research on metabolic diseases, tumors, and cancer.
- Livestock Field: Studies on gut and rumen microorganisms and their roles in animal health and digestion.
- Agricultural Field: Research on plant-microbe interactions and the impact of agricultural practices on soil microbiomes.
- Environmental Field: Research on smog treatment, wastewater management, petroleum degradation, acid mine drainage, and marine environments.
- Bioenergy: Exploration of functional strains, gene discovery, and engineered microorganisms.
- Extreme Environments: Study of microbes in extreme conditions.
Metagenomic shotgun sequencing can profoundly expedite research across various domains:
(i) unraveling the genetic diversity of host-associated microbes;
(ii) elucidating the functional heterogeneity within microbial consortia;
(iii) advancing gene prediction and annotation techniques;
(iv) deciphering the intricate host-microbe interactions;
(v) uncovering microbiota-based disease mechanisms.
Our platform is equipped to assist a diverse clientele, encompassing fields such as scientific inquiry, medical research, agricultural studies, bioengineering, the pharmaceutical industry, and environmental remediation.



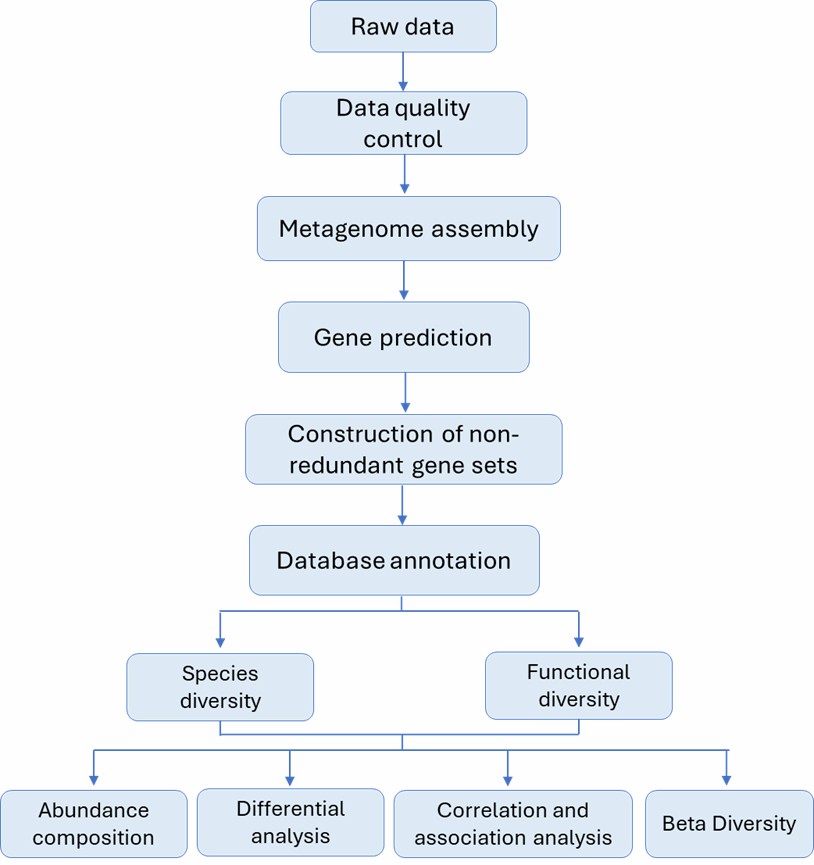

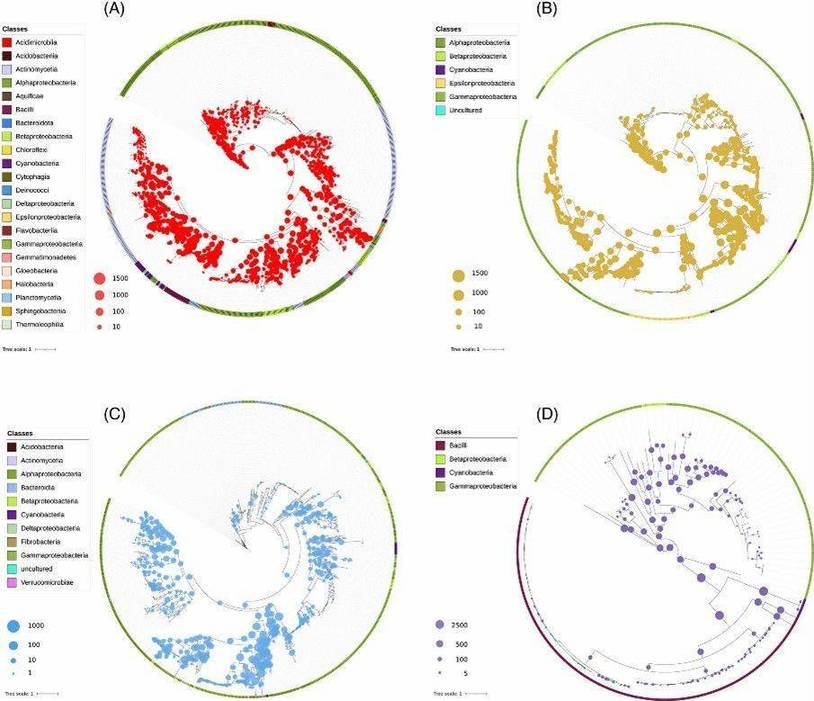 Fig 1. Phylogenetic placements of the predicted proteins of each metagenome with respect to the reference bases of each enzyme: (A) PhoD, (B) PhoX, (C) Nsap-A and (D) BPP.
Fig 1. Phylogenetic placements of the predicted proteins of each metagenome with respect to the reference bases of each enzyme: (A) PhoD, (B) PhoX, (C) Nsap-A and (D) BPP.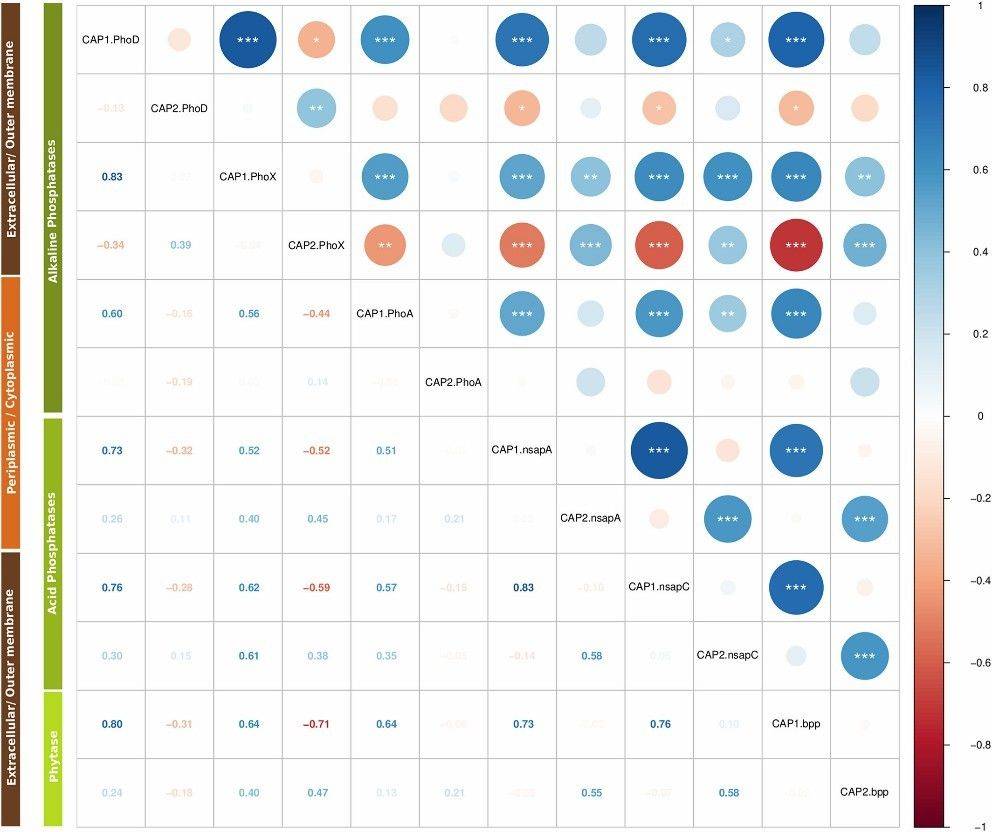 Fig 2. Correlation matrix of KR-CAP axes.
Fig 2. Correlation matrix of KR-CAP axes.