Service Specifications
Our Bacterial 16S rRNA Sequencing Service
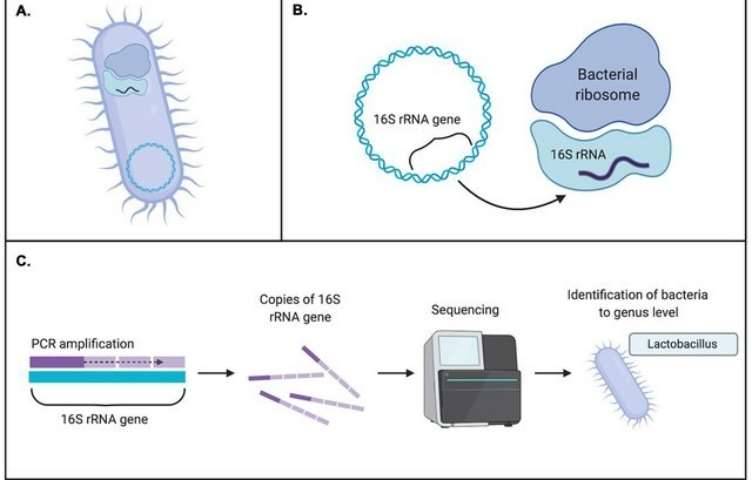
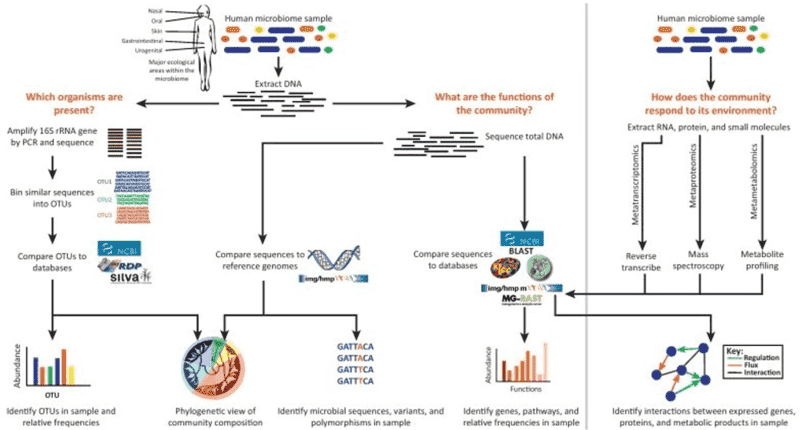
16S rRNA sequencing typically involves the selective PCR amplification of the 16S rRNA gene using specific primers, followed by second-generation or third-generation sequencing. NGS can be performed using platforms such as Illumina MiSeq/HiSeq, Roche 454, or Ion Torrent, while third-generation sequencing leverages PacBio SMRT or Nanopore technologies. Utilizing robust bioinformatics tools, we can conduct essential sequence processing, diversity analysis, taxonomic assignment, and community comparisons. Our powerful 16S rRNA analysis pipeline aligns sequences for phylogenetic assignment using three popular databases: Silva, Green Genes, and the Ribosomal Database Project (RDP).
Bacterial 16S rRNA sequencing aids in determining microbial diversity, abundance, and potential microbial functions. It plays a critical role in identifying bacterial communities across global health and disease populations, contributing significantly to disease diagnosis, biomarker discovery, and target identification. Beyond medical applications, 16S rRNA sequencing is also employed in environmental conservation, agricultural production, oil exploration, and industrial manufacturing.
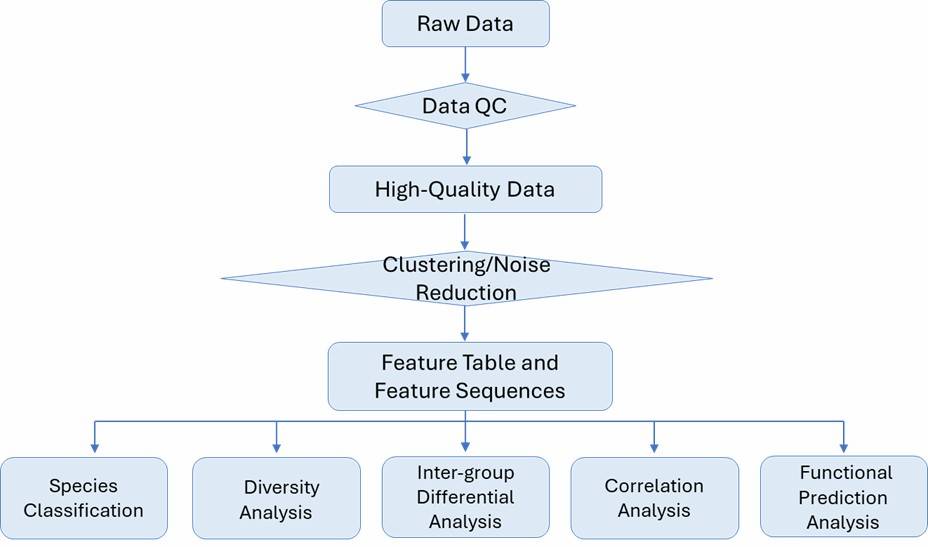
Bacterial 16S rRNA Sequencing Workflow
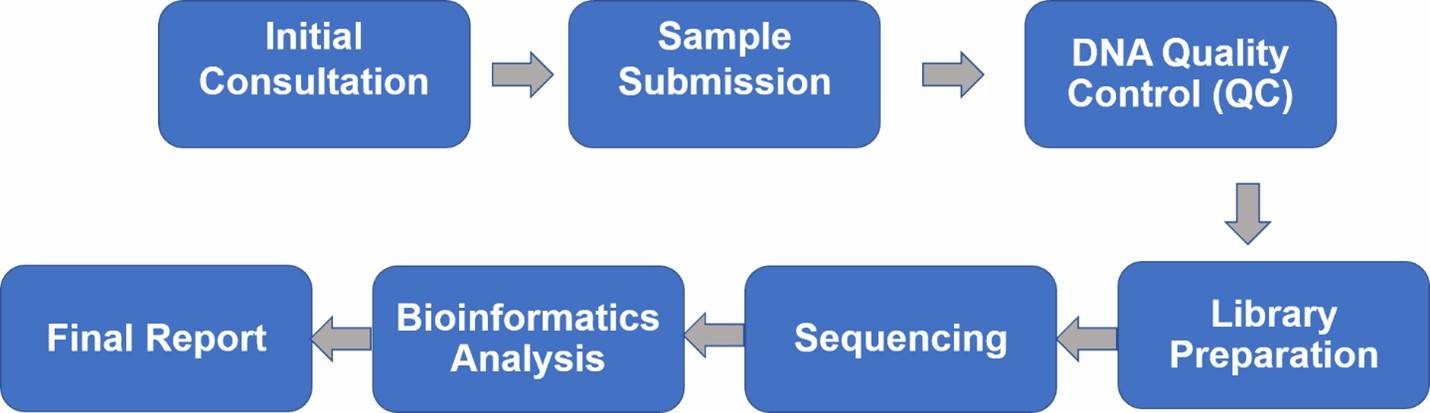
1. Initial Consultation: Begin with an essential consultation with our experts to discuss your samples and project objectives, ensuring well-defined goals and clear expectations.
2. Sample Submission: We provide detailed guidelines on sample handling, processing, and transportation, tailored to the specific sample type and requirements.
3. DNA Quality Control (QC): Prior to library preparation, we rigorously assess the DNA for quantity, quality, and purity.
4. Library Preparation: We construct diverse 16S rRNA libraries conducive to various sequencing platforms, tailored to your project needs.
5. Sequencing: Our comprehensive 16S rRNA sequencing services utilize cutting-edge second- and third-generation sequencing technologies.
6. Bioinformatics Analysis: We offer thorough bioinformatics analysis, encompassing raw data processing, diversity analysis, taxonomic assignments, and community comparisons.
7. Final Report: Delivering a comprehensive and timely final report, we ensure all findings are meticulously documented and accessible.
Technical Parameters:
| Sequencing Platform |
Sequencing Strategy |
Data Volume |
| HiSeq/MiSeq |
PE250 |
Depending on the specific project requirements, not less than the contracted data volume |
Bioinformatics Analysis
Our bioinformatics analysis pipeline is flexible to your needs.
| Pipeline |
Analysis Content |
| Basic data processing |
Filtering and trimming of poor-quality sequence |
| Taxonomic assignment |
Operation Taxonomic Unit (OTU) clustering and species annotation |
| Diversity analysis |
Classification and abundance analysis of single species |
| Community comparisons |
Measure the difference in bacterial community composition among different samples, and conduct network analysis and correlation analysis |
| Evolutionary analysis |
Construction of phylogenetic tree |
Note: The above content includes only a portion of the bioinformatics analysis. For more information or to customize the analysis, please contact us directly.
Sample Requirement
| Sample Type |
Quantity |
Concentration |
OD260/OD280 |
| Genomic DNA |
≥100 ng |
≥1 ng/μl |
1. 8 - 2. 0 |
Note:
1. Ensure the DNA is purified, not degraded.
2. Transport nucleic acid samples with sufficient ice packs or dry ice.
3. Sampling kits: We provide a range of microbial sampling kits for clients, including MicroCollect™ oral sample microbial collection products and MicroCollect™ stool sample collection products.
4. If you wish to obtain more accurate and detailed information regarding sample requirements, please feel free to contact us directly.
Deliverables
- Raw sequencing data
- Trimmed and stitched sequences (FASTA)
- Data QC report
- Custom bioinformatics analysis report


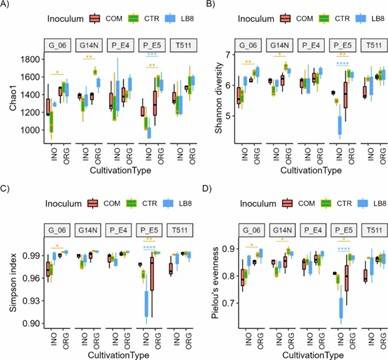 Fig 1. Alpha diversity indices (chao1, Simpson, and Shannon) and Pielou's evenness for different genotypes in the organic and inorganic plots for different inoculums.
Fig 1. Alpha diversity indices (chao1, Simpson, and Shannon) and Pielou's evenness for different genotypes in the organic and inorganic plots for different inoculums.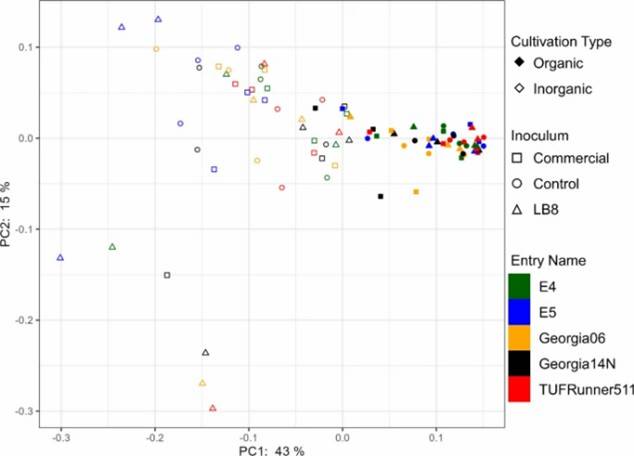 Fig 2. PCoA plot of bacterial community generated by using the weighted UniFrac as distance.
Fig 2. PCoA plot of bacterial community generated by using the weighted UniFrac as distance.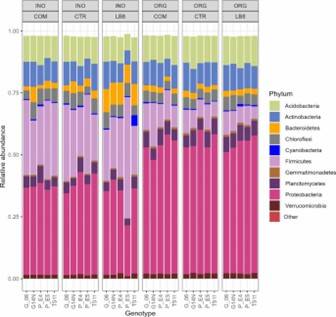 Fig 3. Phylum composition of peanut rhizosphere.
Fig 3. Phylum composition of peanut rhizosphere.